

Computational approaches to interpreting genomic sequence variation
- PMID: 25473426
- PMCID: PMC4254438
- DOI: 10.1186/s13073-014-0087-1
Computational approaches to interpreting genomic sequence variation
Abstract
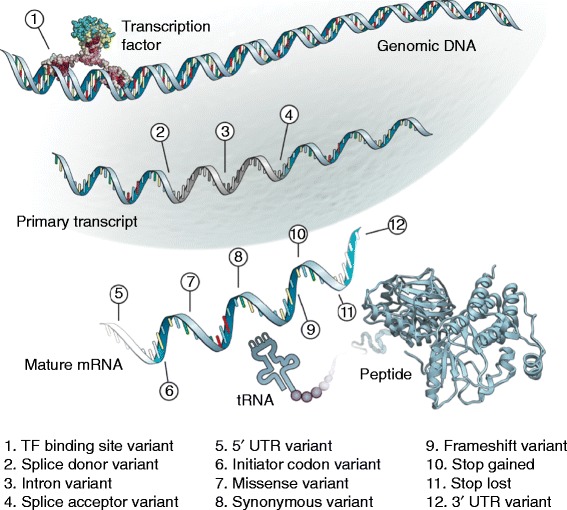
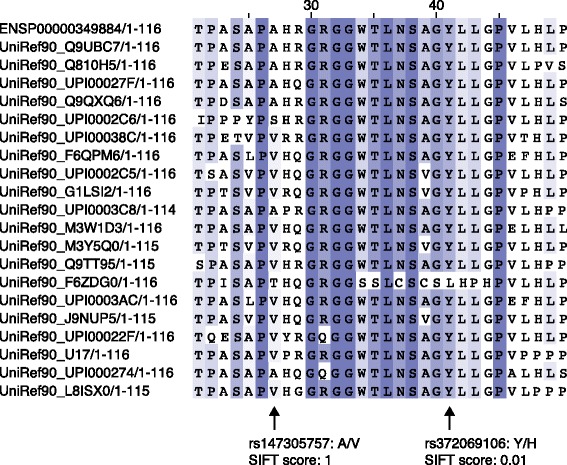
Identifying sequence variants that play a mechanistic role in human disease and other phenotypes is a fundamental goal in human genetics and will be important in translating the results of variation studies. Experimental validation to confirm that a variant causes the biochemical changes responsible for a given disease or phenotype is considered the gold standard, but this cannot currently be applied to the 3 million or so variants expected in an individual genome. This has prompted the development of a wide variety of computational approaches that use several different sources of information to identify functional variation. Here, we review and assess the limitations of computational techniques for categorizing variants according to functional classes, prioritizing variants for experimental follow-up and generating hypotheses about the possible molecular mechanisms to inform downstream experiments. We discuss the main current bioinformatics approaches to identifying functional variation, including widely used algorithms for coding variation such as SIFT and PolyPhen and also novel techniques for interpreting variation across the genome.
Figures

References
-
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. - DOI - PMC - PubMed
-
- Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, Murphy MR, O’Leary NA, Pujar S, Rajput B, Rangwala SH, Riddick LD, Shkeda A, Sun H, Tamez P, Tully RE, Wallin C, Webb D, Weber J, Wu W, DiCuccio M, Kitts P, Maglott DR, Murphy TD, Ostell JM. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. - DOI - PMC - PubMed
-
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2013;133:1–9. doi: 10.1007/s00439-013-1358-4. - DOI - PMC - PubMed
-
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
