

Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations
- PMID: 25473427
- PMCID: PMC4254423
- DOI: 10.1186/s13073-014-0091-5
Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations
Abstract
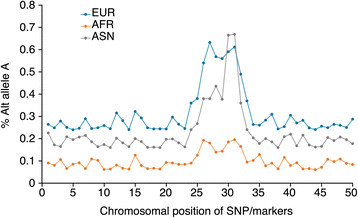
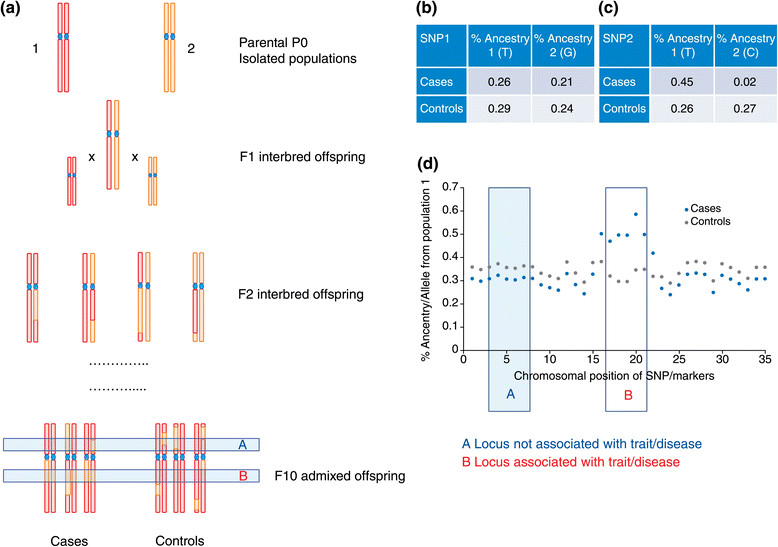
Genome-wide association studies (GWASs) are the method most often used by geneticists to interrogate the human genome, and they provide a cost-effective way to identify the genetic variants underpinning complex traits and diseases. Most initial GWASs have focused on genetically homogeneous cohorts from European populations given the limited availability of ethnic minority samples and so as to limit population stratification effects. Transethnic studies have been invaluable in explaining the heritability of common quantitative traits, such as height, and in examining the genetic architecture of complex diseases, such as type 2 diabetes. They provide an opportunity for large-scale signal replication in independent populations and for cross-population meta-analyses to boost statistical power. In addition, transethnic GWASs enable prioritization of candidate genes, fine-mapping of functional variants, and potentially identification of SNPs associated with disease risk in admixed populations, by taking advantage of natural differences in genomic linkage disequilibrium across ethnically diverse populations. Recent efforts to assess the biological function of variants identified by GWAS have highlighted the need for large-scale replication, meta-analyses and fine-mapping across worldwide populations of ethnically diverse genetic ancestries. Here, we review recent advances and new approaches that are important to consider when performing, designing or interpreting transethnic GWASs, and we highlight existing challenges, such as the limited ability to handle heterogeneity in linkage disequilibrium across populations and limitations in dissecting complex architectures, such as those found in recently admixed populations.
Figures
References
-
- NHGRI: Catalog of published genome-wide association studies. In , [http://www.genome.gov/gwastudies/] - PMC - PubMed
-
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TFC, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
