

Extensive error in the number of genes inferred from draft genome assemblies
- PMID: 25474019
- PMCID: PMC4256071
- DOI: 10.1371/journal.pcbi.1003998
Extensive error in the number of genes inferred from draft genome assemblies
Abstract
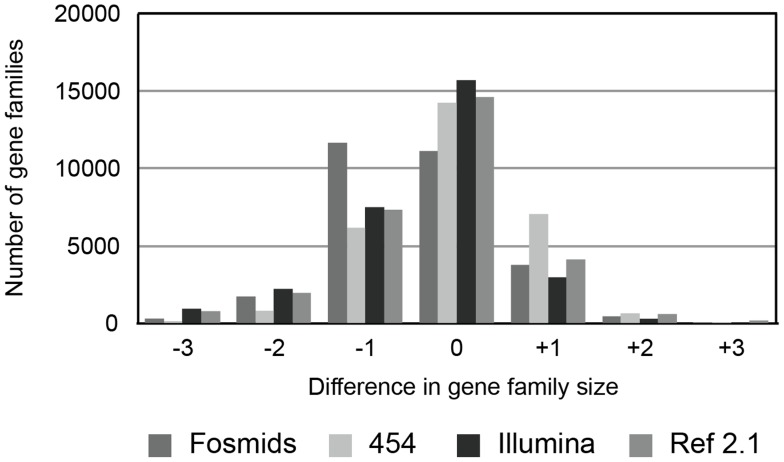
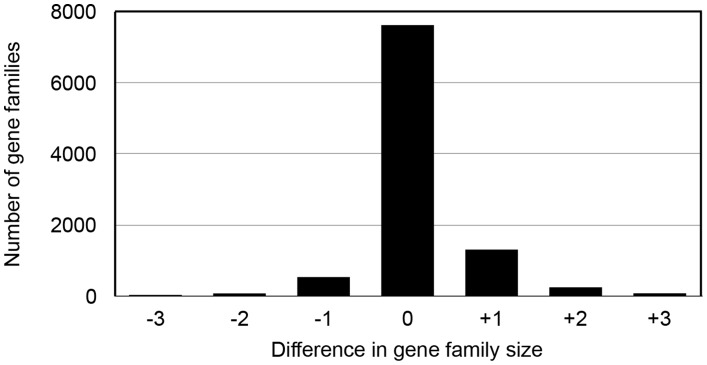
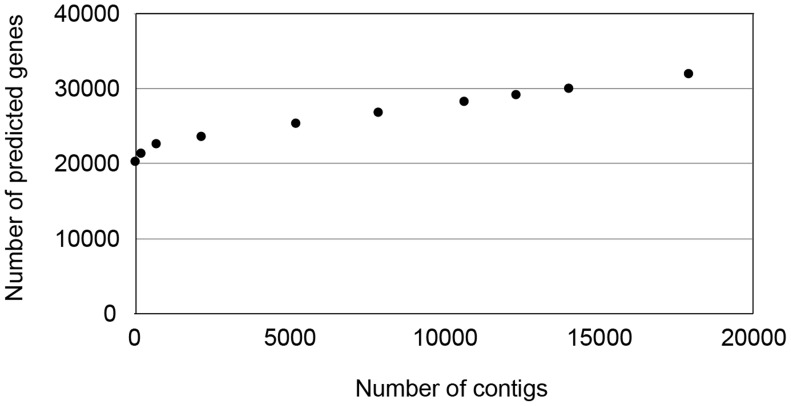
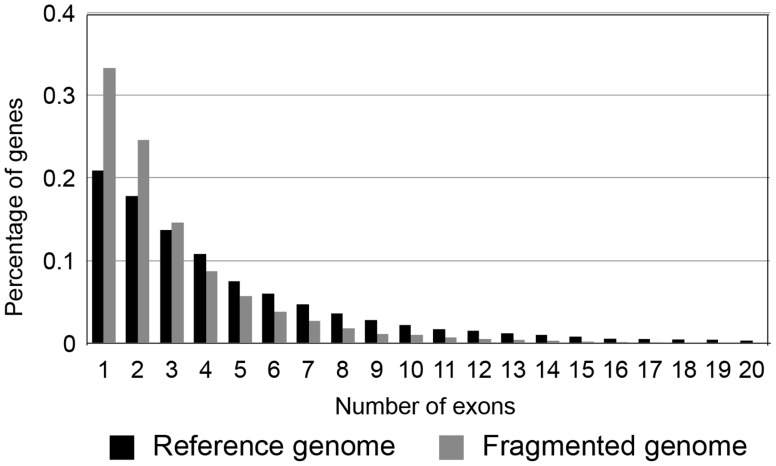
Current sequencing methods produce large amounts of data, but genome assemblies based on these data are often woefully incomplete. These incomplete and error-filled assemblies result in many annotation errors, especially in the number of genes present in a genome. In this paper we investigate the magnitude of the problem, both in terms of total gene number and the number of copies of genes in specific families. To do this, we compare multiple draft assemblies against higher-quality versions of the same genomes, using several new assemblies of the chicken genome based on both traditional and next-generation sequencing technologies, as well as published draft assemblies of chimpanzee. We find that upwards of 40% of all gene families are inferred to have the wrong number of genes in draft assemblies, and that these incorrect assemblies both add and subtract genes. Using simulated genome assemblies of Drosophila melanogaster, we find that the major cause of increased gene numbers in draft genomes is the fragmentation of genes onto multiple individual contigs. Finally, we demonstrate the usefulness of RNA-Seq in improving the gene annotation of draft assemblies, largely by connecting genes that have been fragmented in the assembly process.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Floudas D, Binder M, Riley R, Barry K, Blanchette RA, et al. (2012) The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336: 1715–1719. - PubMed
-
- Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M (2008) Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster . Science 320: 1629–1631. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
