

Association mapping across numerous traits reveals patterns of functional variation in maize
- PMID: 25474422
- PMCID: PMC4256217
- DOI: 10.1371/journal.pgen.1004845
Association mapping across numerous traits reveals patterns of functional variation in maize
Abstract
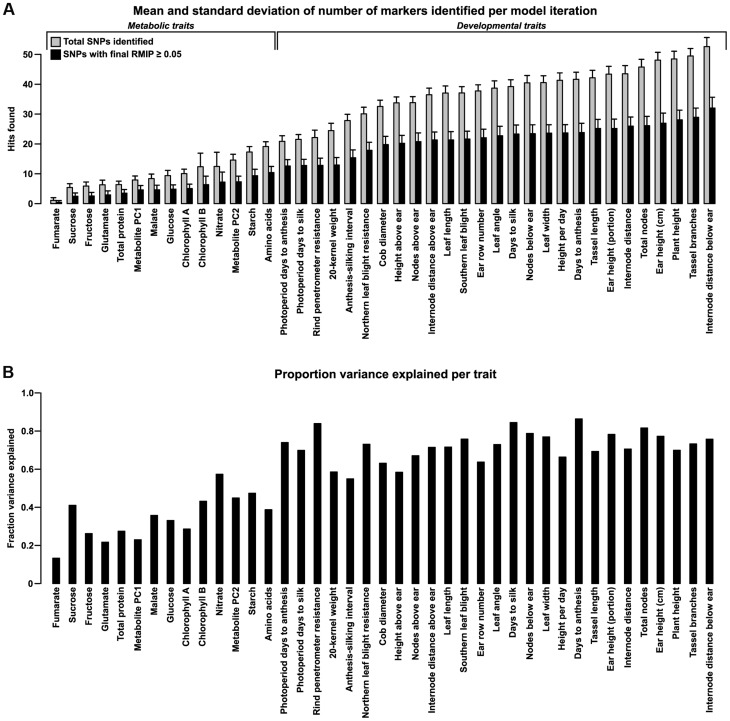
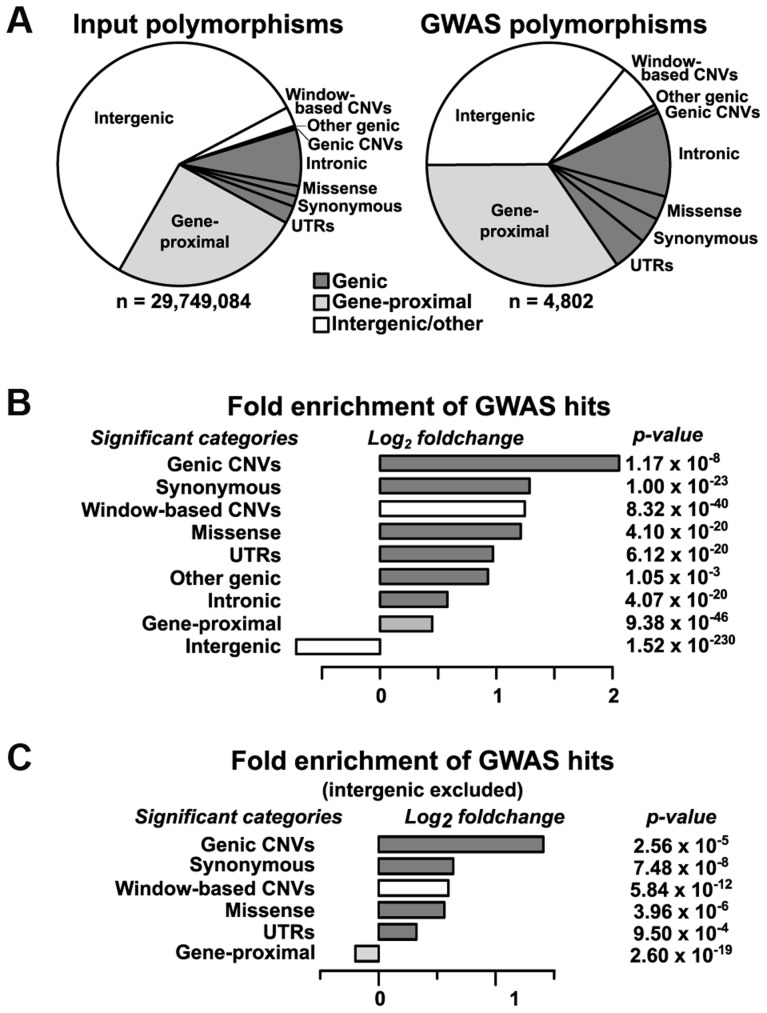
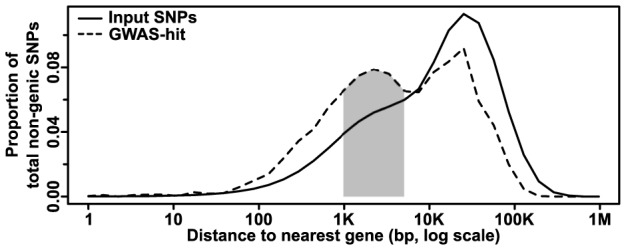
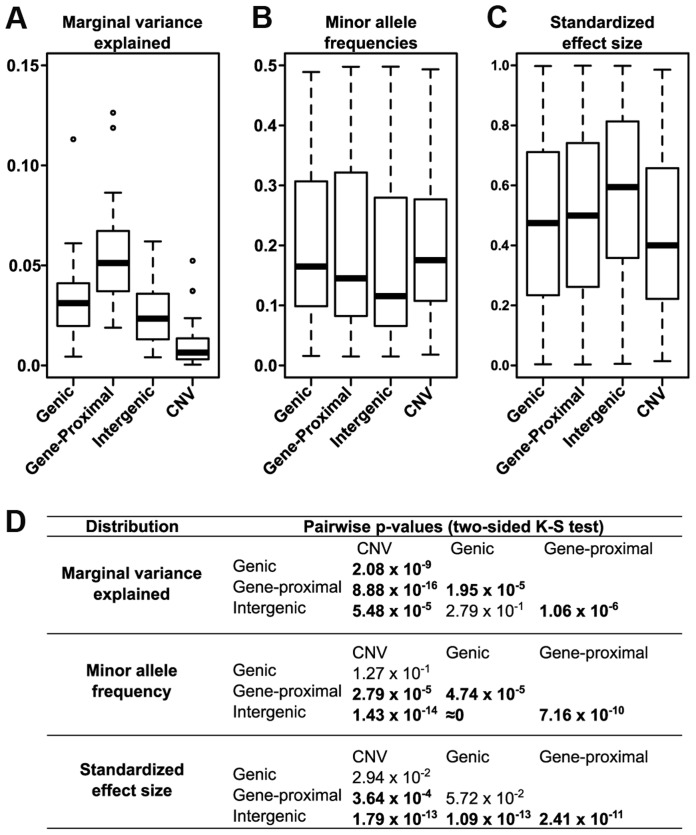
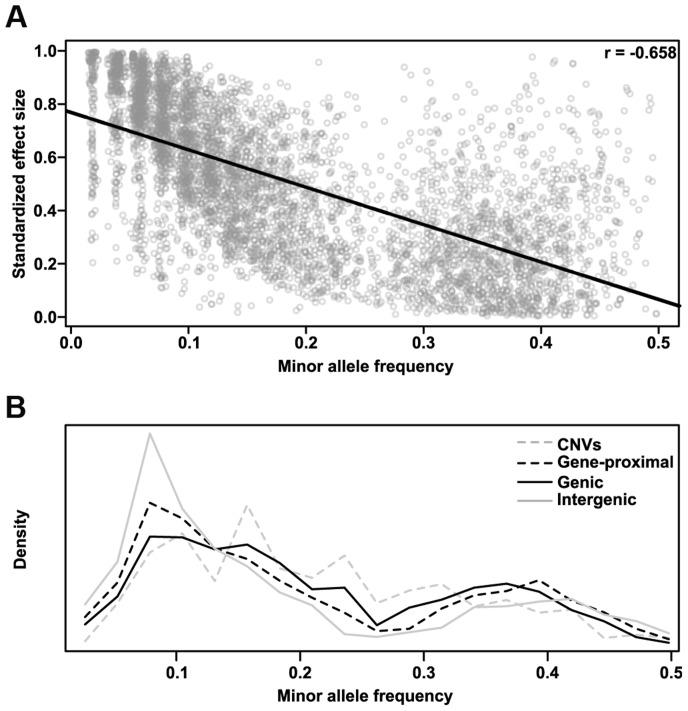
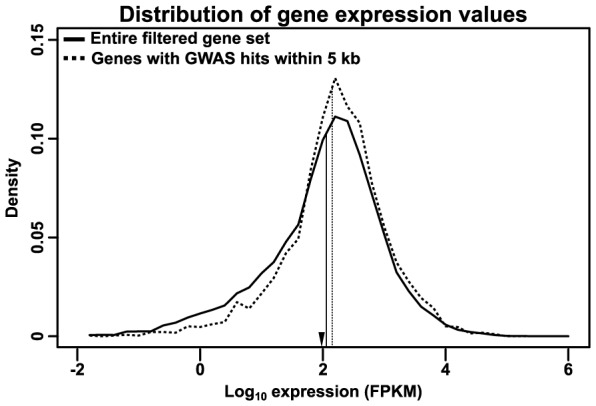
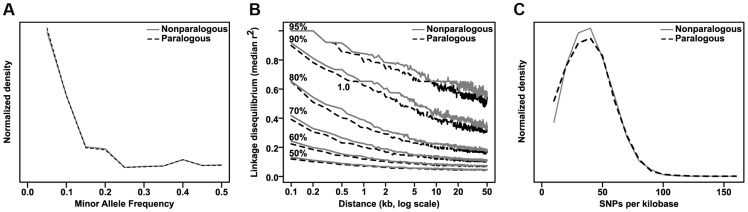
Phenotypic variation in natural populations results from a combination of genetic effects, environmental effects, and gene-by-environment interactions. Despite the vast amount of genomic data becoming available, many pressing questions remain about the nature of genetic mutations that underlie functional variation. We present the results of combining genome-wide association analysis of 41 different phenotypes in ∼ 5,000 inbred maize lines to analyze patterns of high-resolution genetic association among of 28.9 million single-nucleotide polymorphisms (SNPs) and ∼ 800,000 copy-number variants (CNVs). We show that genic and intergenic regions have opposite patterns of enrichment, minor allele frequencies, and effect sizes, implying tradeoffs among the probability that a given polymorphism will have an effect, the detectable size of that effect, and its frequency in the population. We also find that genes tagged by GWAS are enriched for regulatory functions and are ∼ 50% more likely to have a paralog than expected by chance, indicating that gene regulation and gene duplication are strong drivers of phenotypic variation. These results will likely apply to many other organisms, especially ones with large and complex genomes like maize.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, et al. (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308: 419–421. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
