

Semaphorin3a promotes advanced diabetic nephropathy
- PMID: 25475434
- PMCID: PMC4407856
- DOI: 10.2337/db14-0719
Semaphorin3a promotes advanced diabetic nephropathy
Abstract
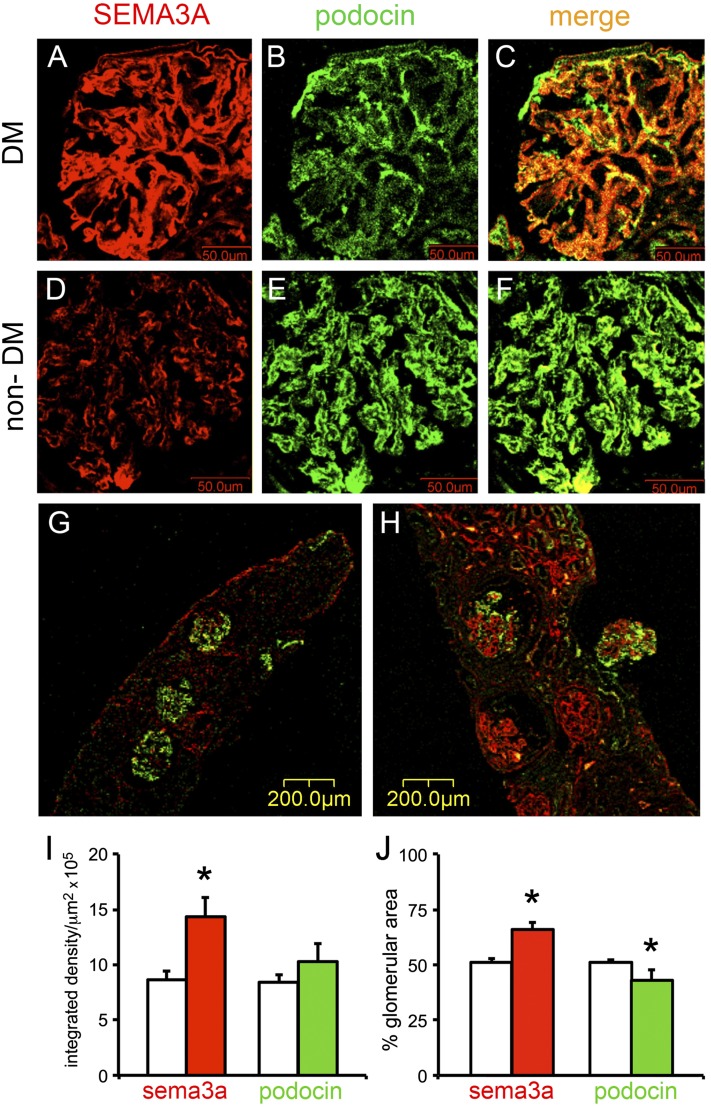
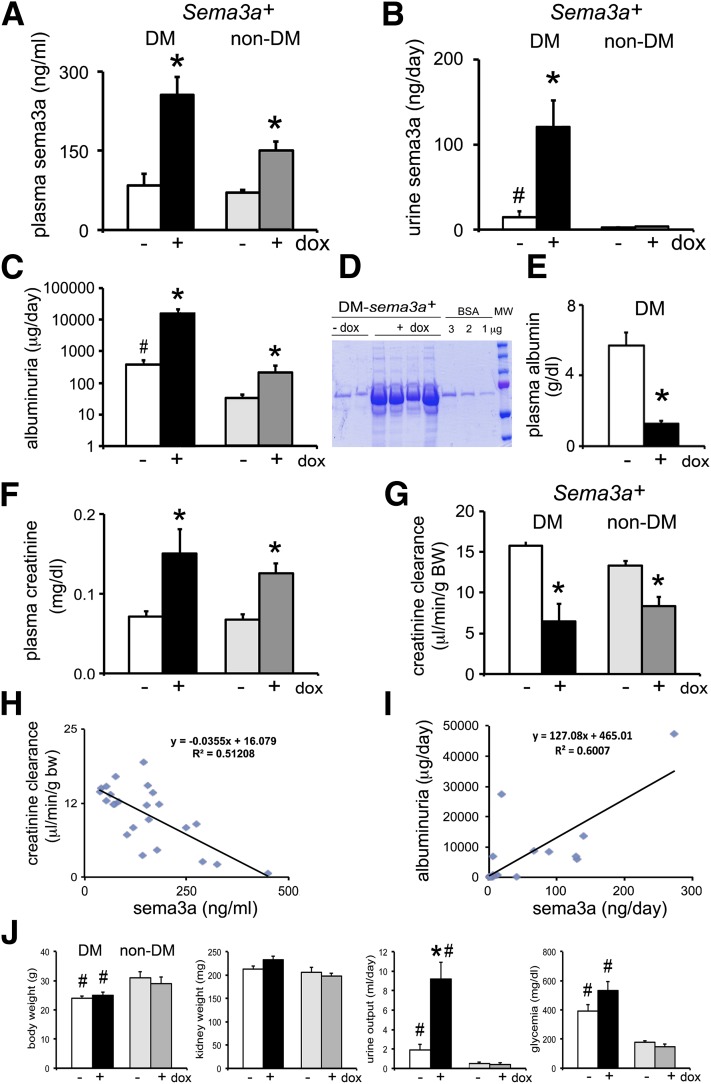
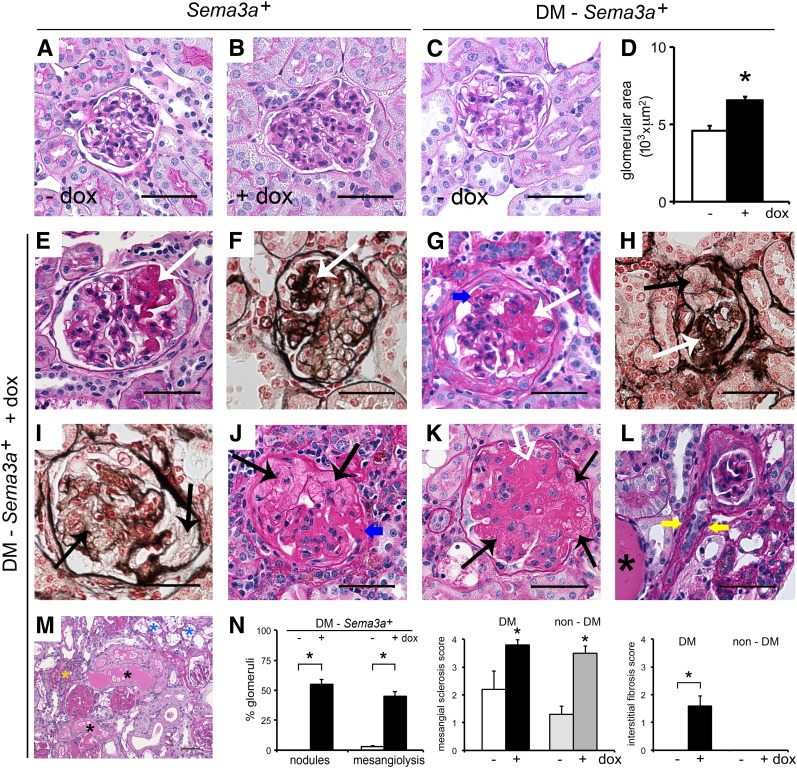
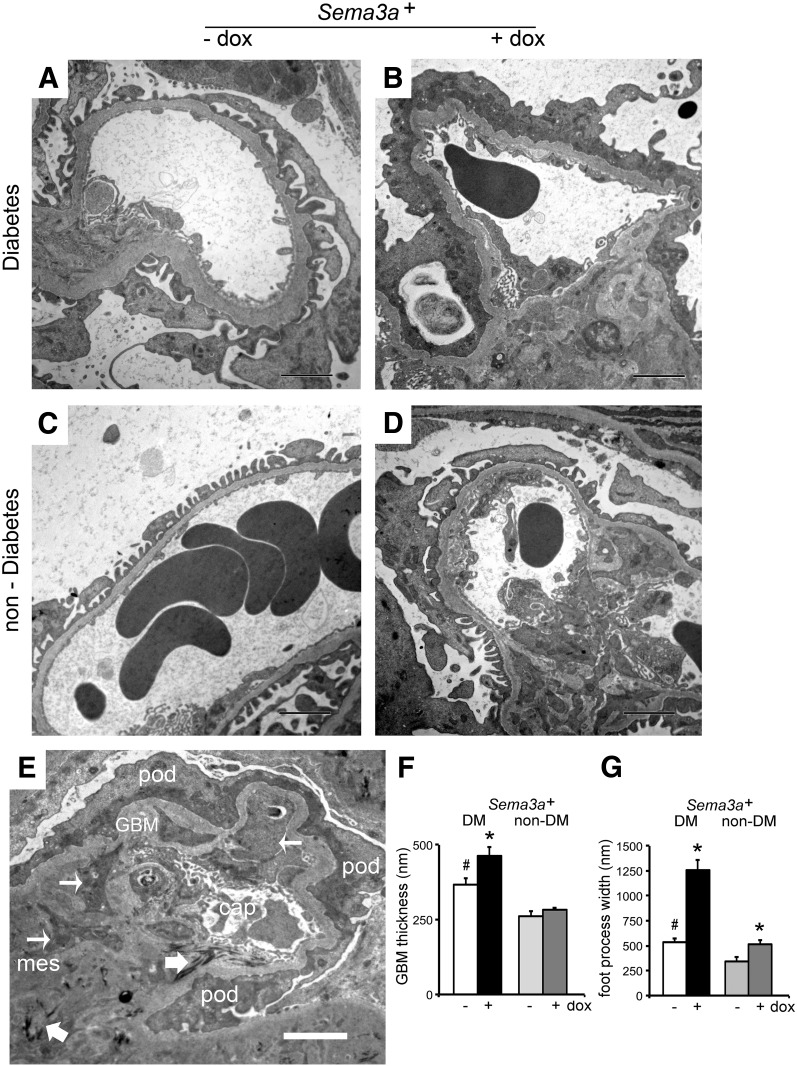
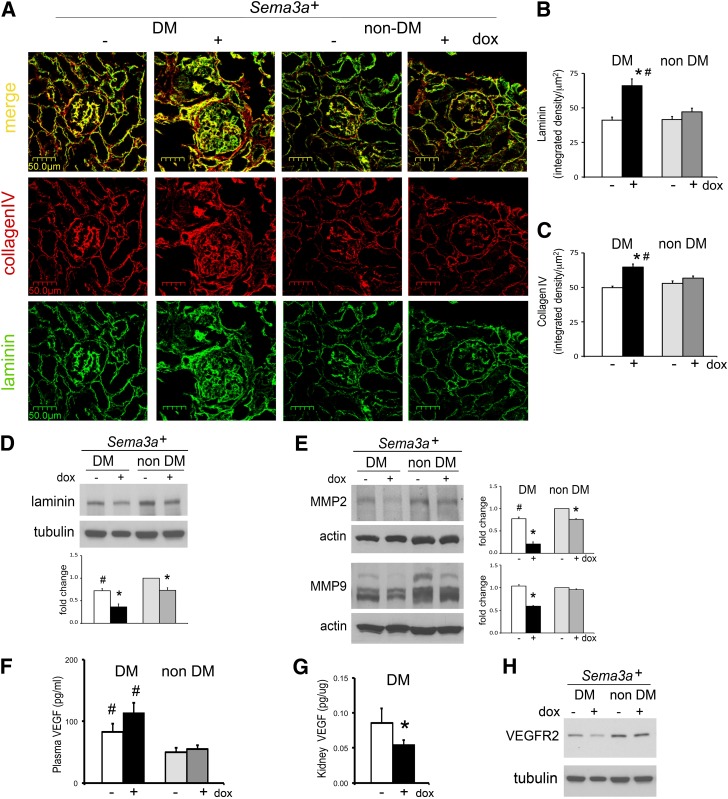
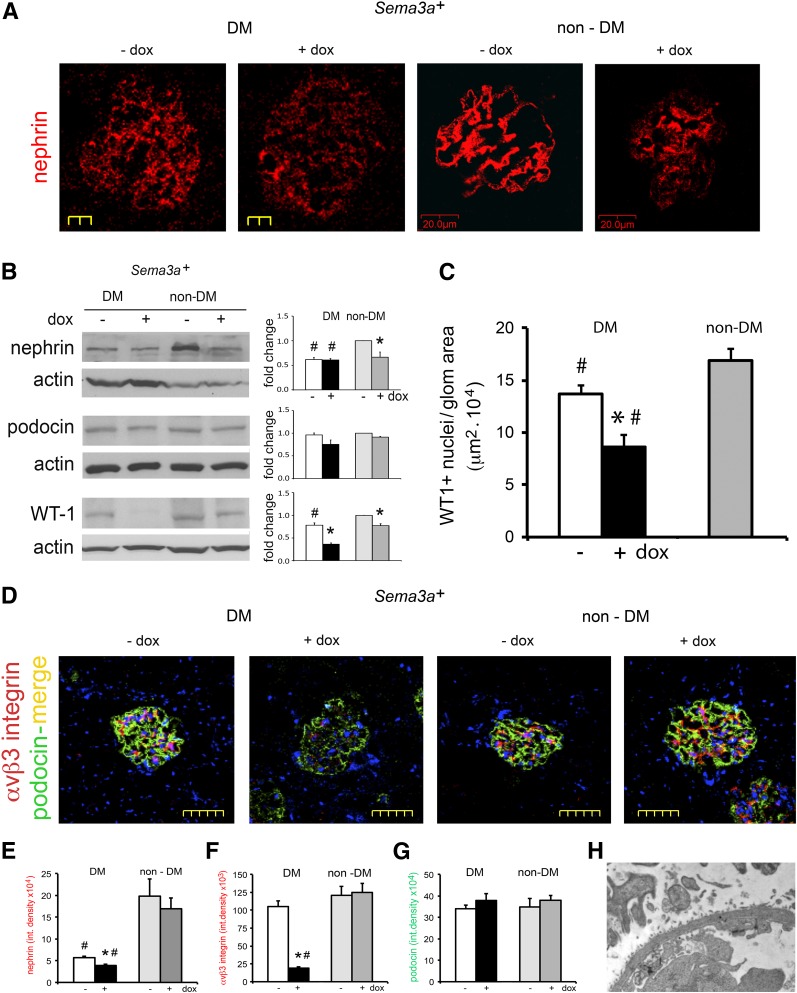
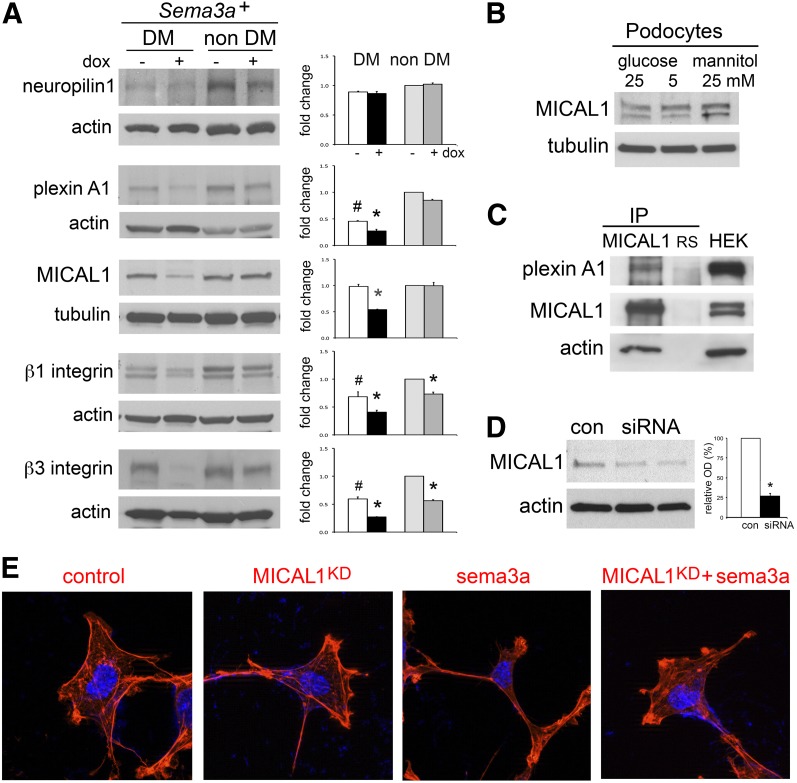
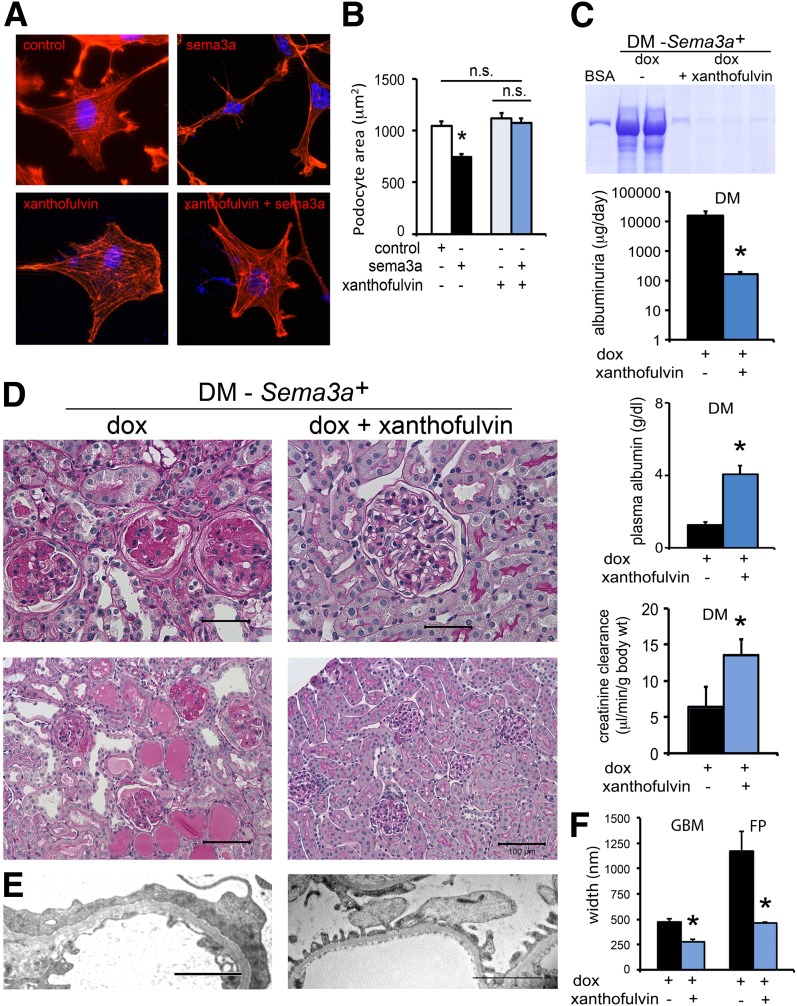
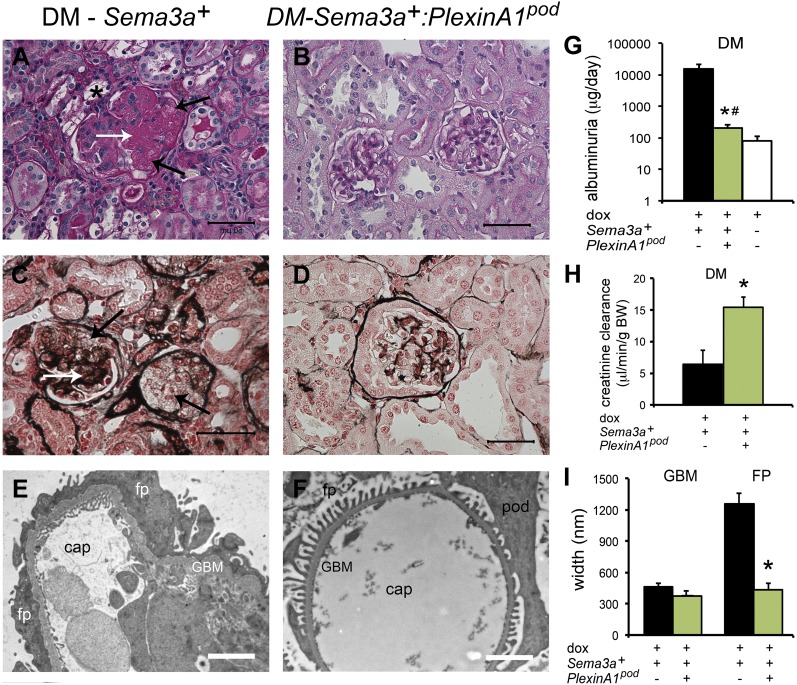
The onset of diabetic nephropathy (DN) is highlighted by glomerular filtration barrier abnormalities. Identifying pathogenic factors and targetable pathways driving DN is crucial to developing novel therapies and improving the disease outcome. Semaphorin3a (sema3a) is a guidance protein secreted by podocytes. Excess sema3a disrupts the glomerular filtration barrier. Here, using immunohistochemistry, we show increased podocyte SEMA3A in renal biopsies from patients with advanced DN. Using inducible, podocyte-specific Sema3a gain-of-function (Sema3a(+)) mice made diabetic with streptozotocin, we demonstrate that sema3a is pathogenic in DN. Diabetic Sema3a(+) mice develop massive proteinuria, renal insufficiency, and extensive nodular glomerulosclerosis, mimicking advanced DN in humans. In diabetic mice, Sema3a(+) exacerbates laminin and collagen IV accumulation in Kimmelstiel-Wilson-like glomerular nodules and causes diffuse podocyte foot process effacement and F-actin collapse via nephrin, αvβ3 integrin, and MICAL1 interactions with plexinA1. MICAL1 knockdown and sema3a inhibition render podocytes not susceptible to sema3a-induced shape changes, indicating that MICAL1 mediates sema3a-induced podocyte F-actin collapse. Moreover, sema3a binding inhibition or podocyte-specific plexinA1 deletion markedly ameliorates albuminuria and abrogates renal insufficiency and the diabetic nodular glomerulosclerosis phenotype of diabetic Sema3a(+) mice. Collectively, these findings indicate that excess sema3a promotes severe diabetic nephropathy and identifies novel potential therapeutic targets for DN.
© 2015 by the American Diabetes Association. Readers may use this article as long as the work is properly cited, the use is educational and not for profit, and the work is not altered.
Figures
References
-
- Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol 2013;9:713–723 - PubMed
-
- Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev 2013;93:137–188 - PubMed
-
- Breyer MD. Drug discovery for diabetic nephropathy: trying the leap from mouse to man. Semin Nephrol 2012;32:445–451 - PubMed
-
- Mogensen CE, Christensen CK, Vittinghus E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes 1983;32(Suppl. 2):64–78 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
