

A framework for modelling gene regulation which accommodates non-equilibrium mechanisms
- PMID: 25475875
- PMCID: PMC4288563
- DOI: 10.1186/s12915-014-0102-4
A framework for modelling gene regulation which accommodates non-equilibrium mechanisms
Abstract
Background: Gene regulation has, for the most part, been quantitatively analysed by assuming that regulatory mechanisms operate at thermodynamic equilibrium. This formalism was originally developed to analyse the binding and unbinding of transcription factors from naked DNA in eubacteria. Although widely used, it has made it difficult to understand the role of energy-dissipating, epigenetic mechanisms, such as DNA methylation, nucleosome remodelling and post-translational modification of histones and co-regulators, which act together with transcription factors to regulate gene expression in eukaryotes.
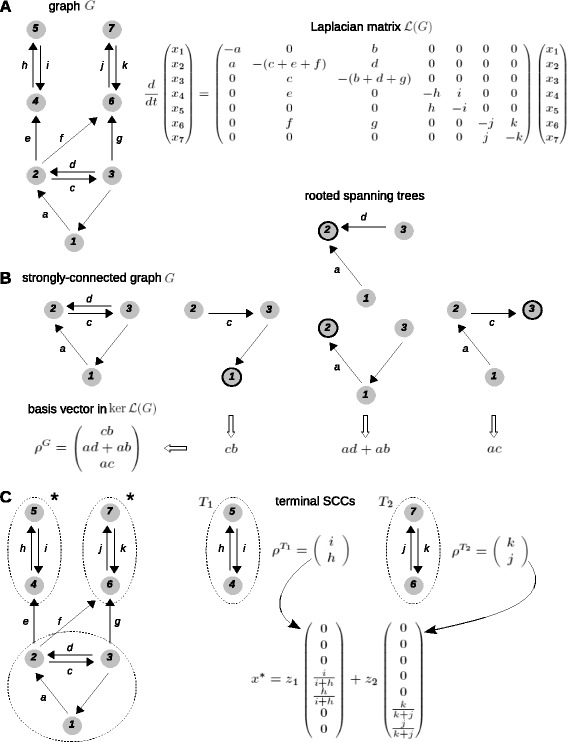
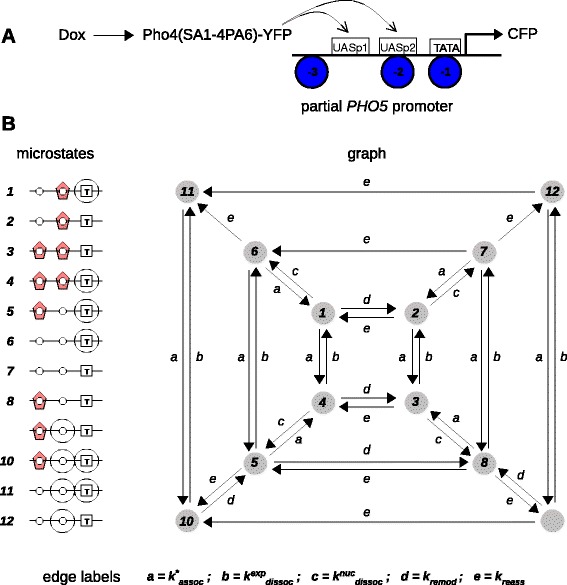
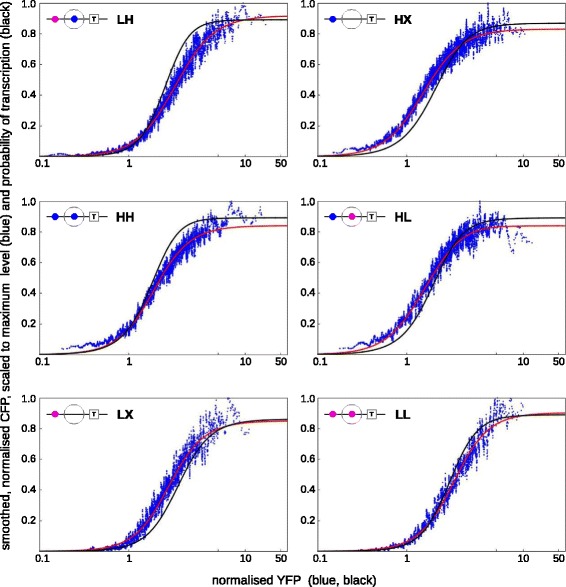
Results: Here, we introduce a graph-based framework that can accommodate non-equilibrium mechanisms. A gene-regulatory system is described as a graph, which specifies the DNA microstates (vertices), the transitions between microstates (edges) and the transition rates (edge labels). The graph yields a stochastic master equation for how microstate probabilities change over time. We show that this framework has broad scope by providing new insights into three very different ad hoc models, of steroid-hormone responsive genes, of inherently bounded chromatin domains and of the yeast PHO5 gene. We find, moreover, surprising complexity in the regulation of PHO5, which has not yet been experimentally explored, and we show that this complexity is an inherent feature of being away from equilibrium. At equilibrium, microstate probabilities do not depend on how a microstate is reached but, away from equilibrium, each path to a microstate can contribute to its steady-state probability. Systems that are far from equilibrium thereby become dependent on history and the resulting complexity is a fundamental challenge. To begin addressing this, we introduce a graph-based concept of independence, which can be applied to sub-systems that are far from equilibrium, and prove that history-dependent complexity can be circumvented when sub-systems operate independently.
Conclusions: As epigenomic data become increasingly available, we anticipate that gene function will come to be represented by graphs, as gene structure has been represented by sequences, and that the methods introduced here will provide a broader foundation for understanding how genes work.
Figures




References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
