

Tumor suppressor ataxia telangiectasia mutated functions downstream of TGF-β1 in orchestrating profibrotic responses
- PMID: 25480384
- PMCID: PMC4396616
- DOI: 10.1096/fj.14-262527
Tumor suppressor ataxia telangiectasia mutated functions downstream of TGF-β1 in orchestrating profibrotic responses
Abstract
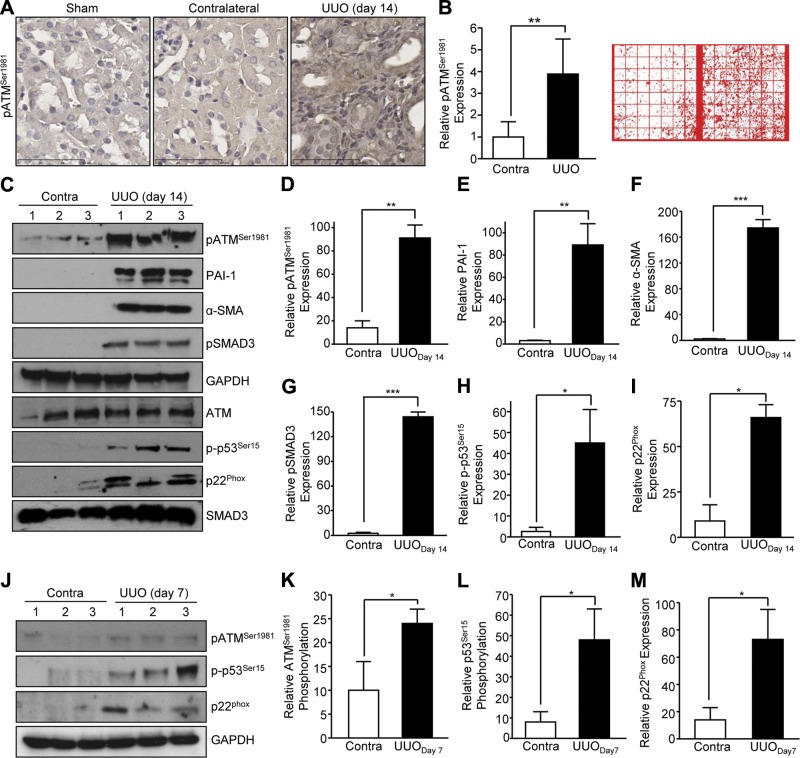
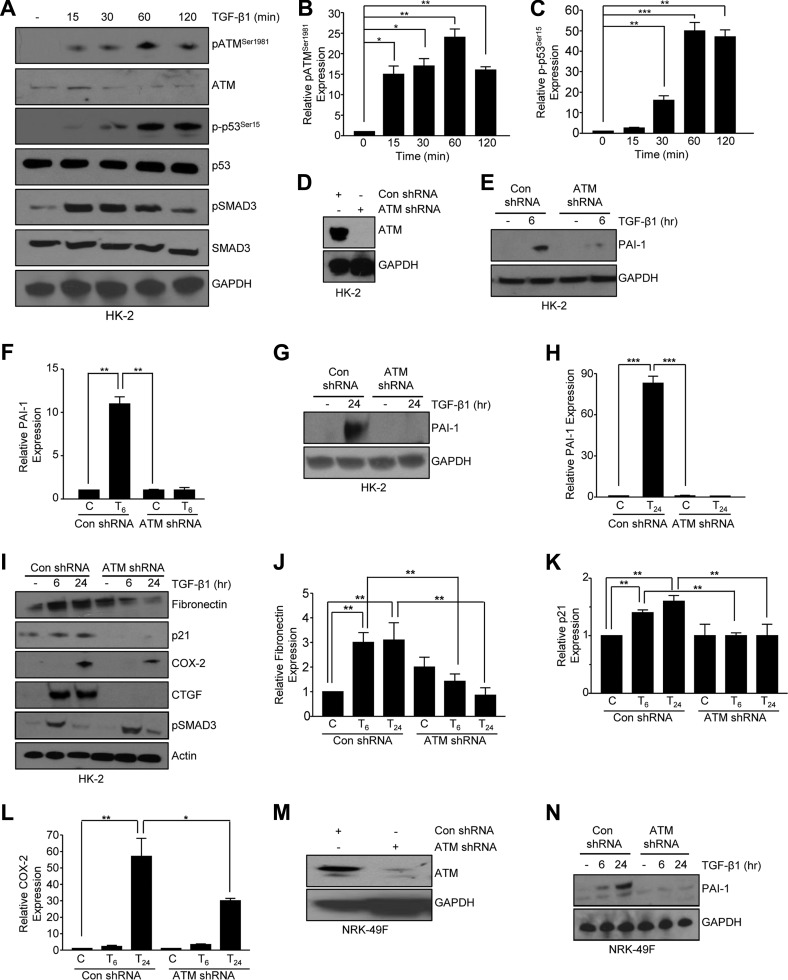
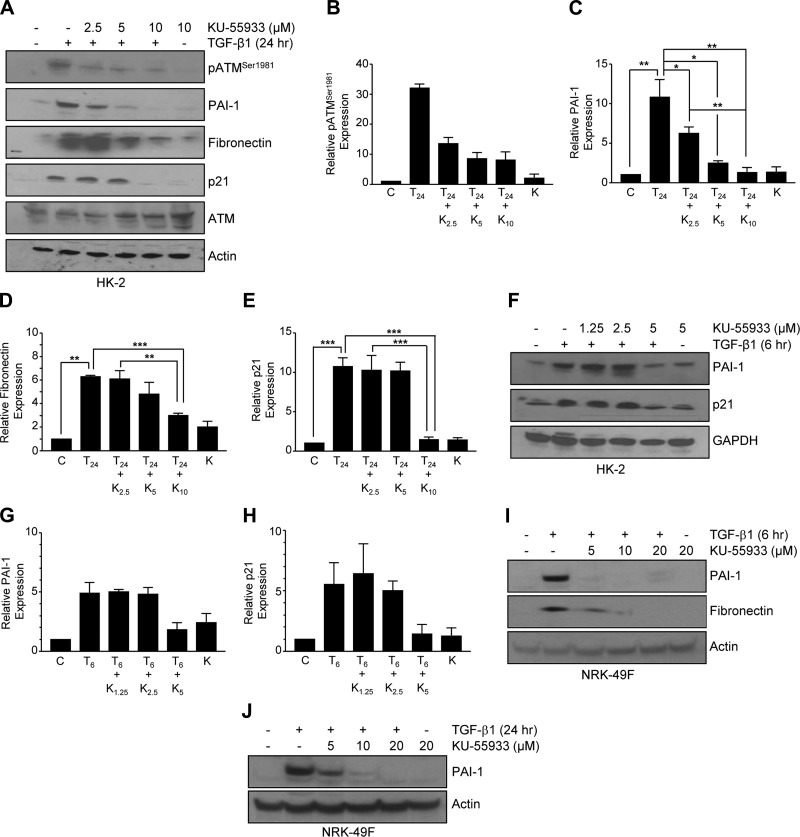
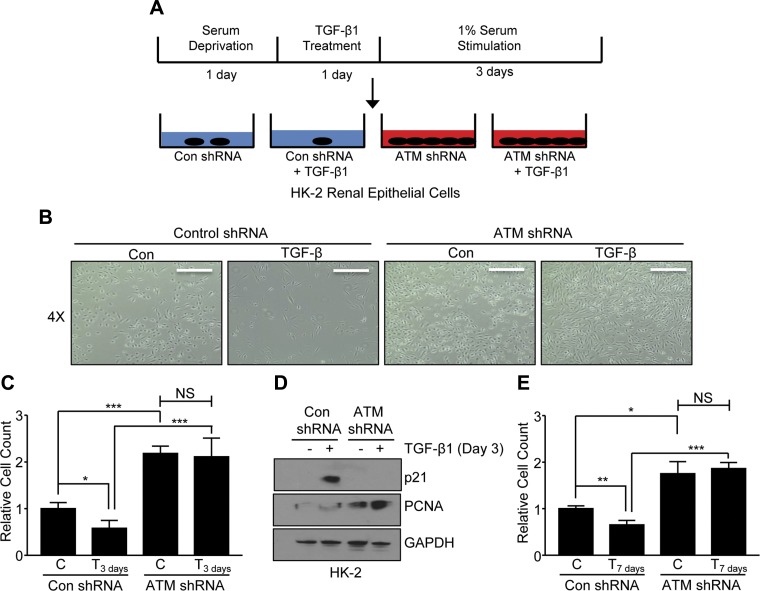
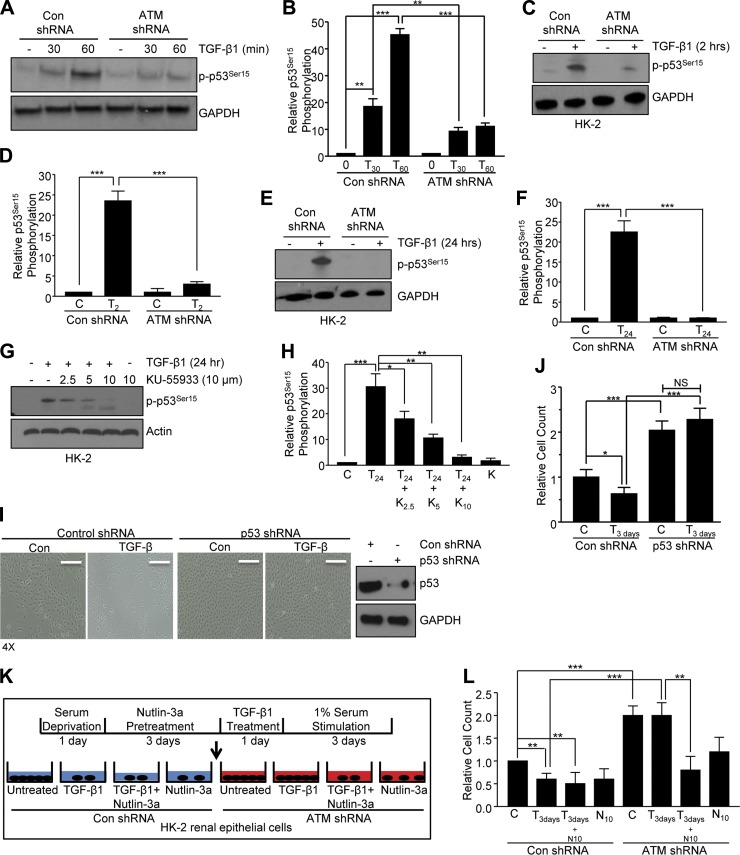
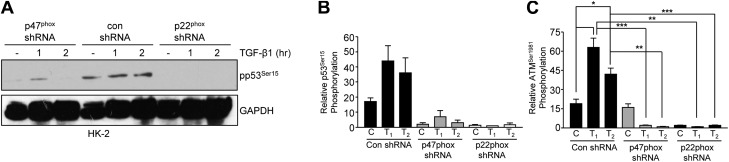
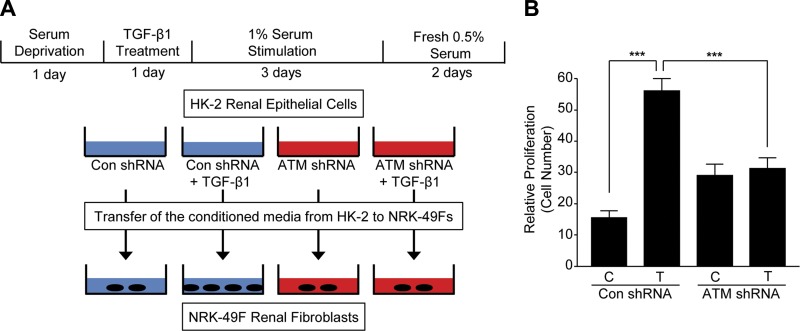
Effective therapy to prevent organ fibrosis, which is associated with more than half of all mortalities, remains elusive. Involvement of tumor suppressor ataxia telangiectasia mutated (ATM) in the TGF-β1 pathway related to renal fibrosis is largely unknown. ATM activation (pATM(Ser1981)) increased 4-fold in the tubulointerstitial region of the unilateral ureteral obstruction-injured kidney in mice correlating with SMAD3 and p53(Ser15) phosphorylation and elevated levels of p22(phox) subunit of the NADPH oxidases (NOXs), and fibrotic markers, plasminogen activator inhibitor-1 (PAI-1), and fibronectin, when compared to contralateral (contra) or sham controls. In fact, ATM is rapidly phosphorylated at Ser(1981) by TGF-β1 stimulation. Stable silencing and pharmacologic inhibition of ATM ablated TGF-β1-induced p53 activation (>95%) and subsequent PAI-1, fibronectin, connective tissue growth factor, and p21 expression in human kidney 2 (HK-2) tubular epithelial cells and normal rat kidney-49 fibroblasts (NRK-49F). ATM or p53 depletion in HK-2 cells, moreover, bypassed TGF-β1-mediated cytostasis evident in control short hairpin RNA-expressing HK-2 cells. Interestingly, stable silencing of NOX subunits, p22(phox) and p47(phox), in HK-2 cells blocked TGF-β1-induced pATM(Ser1981) (>90%) and target gene induction via p53-dependent mechanisms. Furthermore, NRK-49F fibroblast proliferation triggered by conditioned media from TGF-β1-stimulated, control vector-transfected HK-2 cells decreased (∼ 50%) when exposed to conditioned media from ATM-deficient, TGF-β1-treated HK-2 cells. Thus, TGF-β1 promotes NOX-dependent ATM activation leading to p53-mediated fibrotic gene reprogramming and growth arrest in HK-2 cells. Furthermore, TGF-β1/ATM-initiated paracrine factor secretion by dysfunctional renal epithelium promotes interstitial fibroblast growth, suggesting a role of tubular ATM in mediating epithelial-mesenchymal cross-talk highlighting the translational benefit of targeting the NOX/ATM/p53 axis in renal fibrosis.
Keywords: NOX; ROS; kidney fibrosis; p53.
© FASEB.
Figures


Similar articles
-
Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species.Cell Signal. 2013 Nov;25(11):2198-209. doi: 10.1016/j.cellsig.2013.07.007. Epub 2013 Jul 18. Cell Signal. 2013. PMID: 23872073
-
Redox control of p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity.Cell Signal. 2014 Jul;26(7):1427-36. doi: 10.1016/j.cellsig.2014.02.017. Epub 2014 Mar 5. Cell Signal. 2014. PMID: 24613410 Free PMC article.
-
Rac-GTPase promotes fibrotic TGF-β1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways.FASEB J. 2019 Sep;33(9):9797-9810. doi: 10.1096/fj.201802489RR. Epub 2019 May 16. FASEB J. 2019. PMID: 31095421 Free PMC article.
-
TGF-β1-p53 cooperativity regulates a profibrotic genomic program in the kidney: molecular mechanisms and clinical implications.FASEB J. 2019 Oct;33(10):10596-10606. doi: 10.1096/fj.201900943R. Epub 2019 Jul 6. FASEB J. 2019. PMID: 31284746 Free PMC article. Review.
-
TGF-β1/p53 signaling in renal fibrogenesis.Cell Signal. 2018 Mar;43:1-10. doi: 10.1016/j.cellsig.2017.11.005. Epub 2017 Nov 28. Cell Signal. 2018. PMID: 29191563 Free PMC article. Review.
Cited by
-
Co-expression of fibrotic genes in inflammatory bowel disease; A localized event?Front Immunol. 2022 Dec 23;13:1058237. doi: 10.3389/fimmu.2022.1058237. eCollection 2022. Front Immunol. 2022. PMID: 36632136 Free PMC article.
-
Crosstalk between ferroptosis and innate immune in diabetic kidney disease: mechanisms and therapeutic implications.Front Immunol. 2025 Feb 28;16:1505794. doi: 10.3389/fimmu.2025.1505794. eCollection 2025. Front Immunol. 2025. PMID: 40092979 Free PMC article. Review.
-
Unilateral Ureteral Obstruction as a Model to Investigate Fibrosis-Attenuating Treatments.Biomolecules. 2019 Apr 8;9(4):141. doi: 10.3390/biom9040141. Biomolecules. 2019. PMID: 30965656 Free PMC article. Review.
-
The Genomic Response to TGF-β1 Dictates Failed Repair and Progression of Fibrotic Disease in the Obstructed Kidney.Front Cell Dev Biol. 2021 Jul 2;9:678524. doi: 10.3389/fcell.2021.678524. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34277620 Free PMC article. Review.
-
Ataxia-Telangiectasia Mutated Modulation of Carbon Metabolism in Cancer.Front Oncol. 2017 Nov 29;7:291. doi: 10.3389/fonc.2017.00291. eCollection 2017. Front Oncol. 2017. PMID: 29238697 Free PMC article. Review.
References
-
- Friedman S. L., Sheppard D., Duffield J. S., Violette S. (2013) Therapy for fibrotic diseases: nearing the starting line. Sci. Transl. Med. 5, 167sr1. - PubMed
-
- Kisseleva T., Brenner D. A. (2007) Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J. Gastroenterol. Hepatol. 22(Suppl 1), S73–S78 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
