

BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity
- PMID: 25480661
- PMCID: PMC4366655
- DOI: 10.1182/blood-2014-10-603100
BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity
Abstract
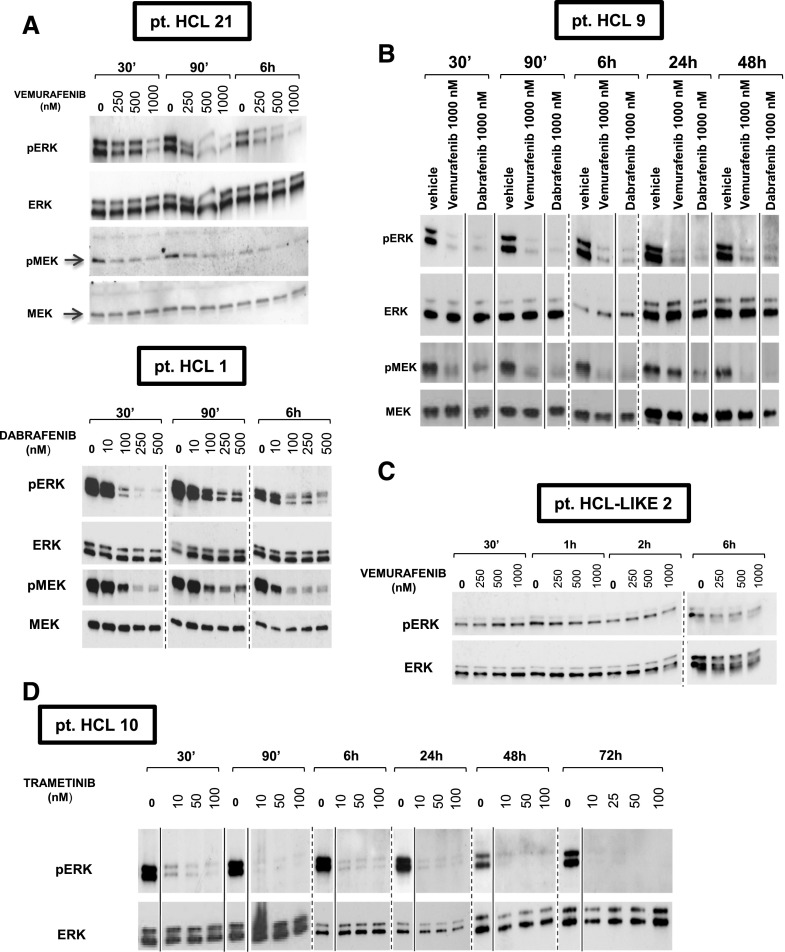
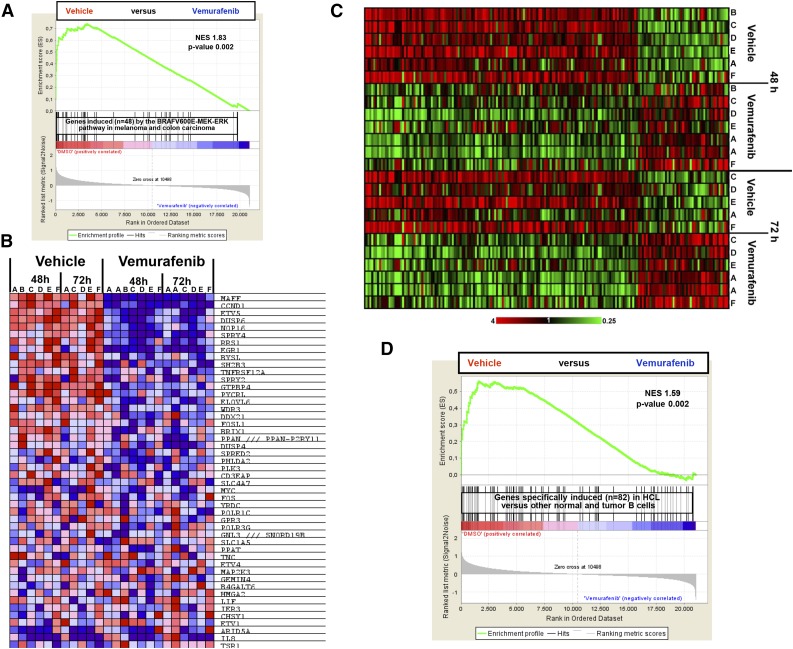
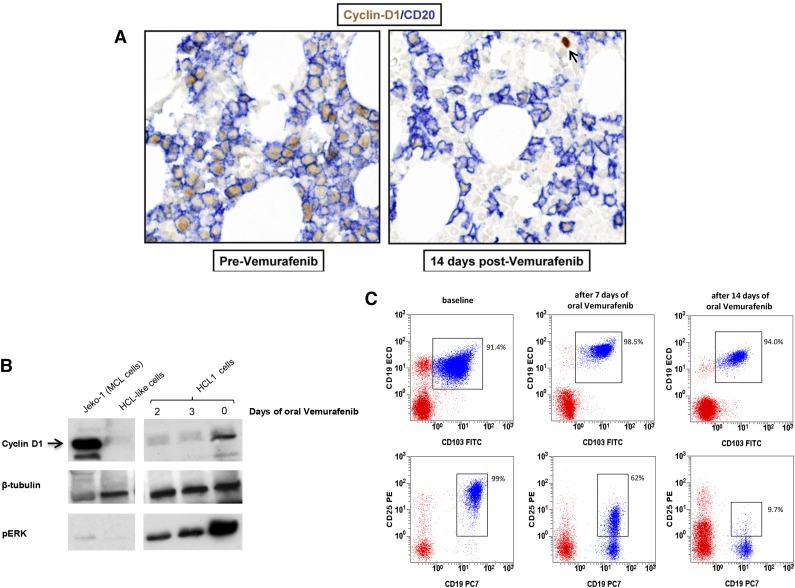
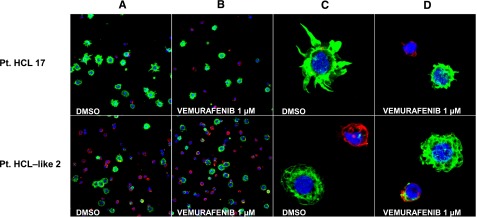
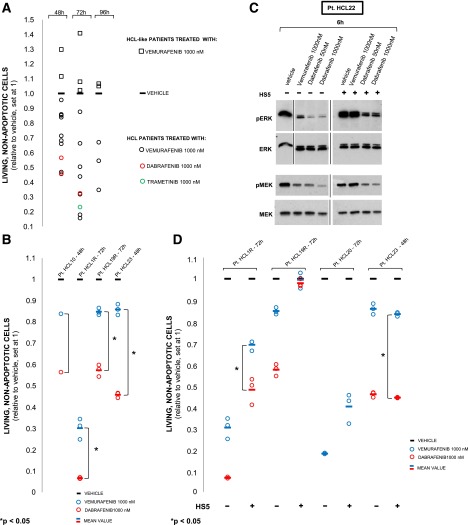
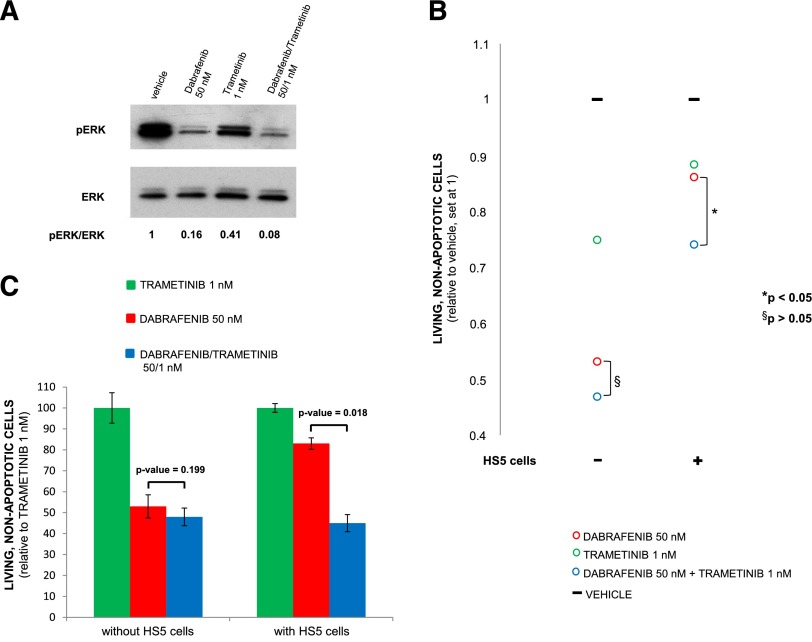
Hairy cell leukemia (HCL) shows unique clinicopathological and biological features. HCL responds well to purine analogs but relapses are frequent and novel therapies are required. BRAF-V600E is the key driver mutation in HCL and distinguishes it from other B-cell lymphomas, including HCL-like leukemias/lymphomas (HCL-variant and splenic marginal zone lymphoma). The kinase-activating BRAF-V600E mutation also represents an ideal therapeutic target in HCL. Here, we investigated the biological and therapeutic importance of the activated BRAF-mitogen-activated protein kinase kinase (MEK)-extracellular signal-regulated kinase (ERK) pathway in HCL by exposing in vitro primary leukemic cells purified from 26 patients to clinically available BRAF (vemurafenib; dabrafenib) or MEK (trametinib) inhibitors. Results were validated in vivo in samples from vemurafenib-treated HCL patients within a phase 2 clinical trial. BRAF and MEK inhibitors caused, specifically in HCL (but not HCL-like) cells, marked MEK/ERK dephosphorylation, silencing of the BRAF-MEK-ERK pathway transcriptional output, loss of the HCL-specific gene expression signature, downregulation of the HCL markers CD25, tartrate-resistant acid phosphatase, and cyclin D1, smoothening of leukemic cells' hairy surface, and, eventually, apoptosis. Apoptosis was partially blunted by coculture with bone marrow stromal cells antagonizing MEK-ERK dephosphorylation. This protective effect could be counteracted by combined BRAF and MEK inhibition. Our results strongly support and inform the clinical use of BRAF and MEK inhibitors in HCL.
© 2015 by The American Society of Hematology.
Figures
Comment in
-
Removing a hair of doubt about BRAF targeting.Blood. 2015 Feb 19;125(8):1199-200. doi: 10.1182/blood-2014-12-616318. Blood. 2015. PMID: 25700421 Free PMC article.
References
-
- Foucar MK, Falini B, Catovsky D, Stein H. In: Hairy Cell Leukemia in WHO Classification of Tumours of Haemapoietic and Lymphoid Tissues. Swerdlow SH, Campo E, Harris NL, et al., editors. Lyon, France: International Agency for Research on Cancer; 2008. pp. 188–190.
-
- Falini B, Tiacci E, Liso A, et al. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet. 2004;363(9424):1869–1870. - PubMed
-
- Tiacci E, Liso A, Piris M, Falini B. Evolving concepts in the pathogenesis of hairy-cell leukaemia. Nat Rev Cancer. 2006;6(6):437–448. - PubMed
-
- Kitagawa Y, Brahmachary M, Tiacci E, Dalla-Favera R, Falini B, Basso K. A microRNA signature specific for hairy cell leukemia and associated with modulation of the MAPK-JNK pathways. Leukemia. 2012;26(12):2564–2567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
