

Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes
- PMID: 25480986
- PMCID: PMC8108008
- DOI: 10.1136/jmedgenet-2014-102856
Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes
Abstract
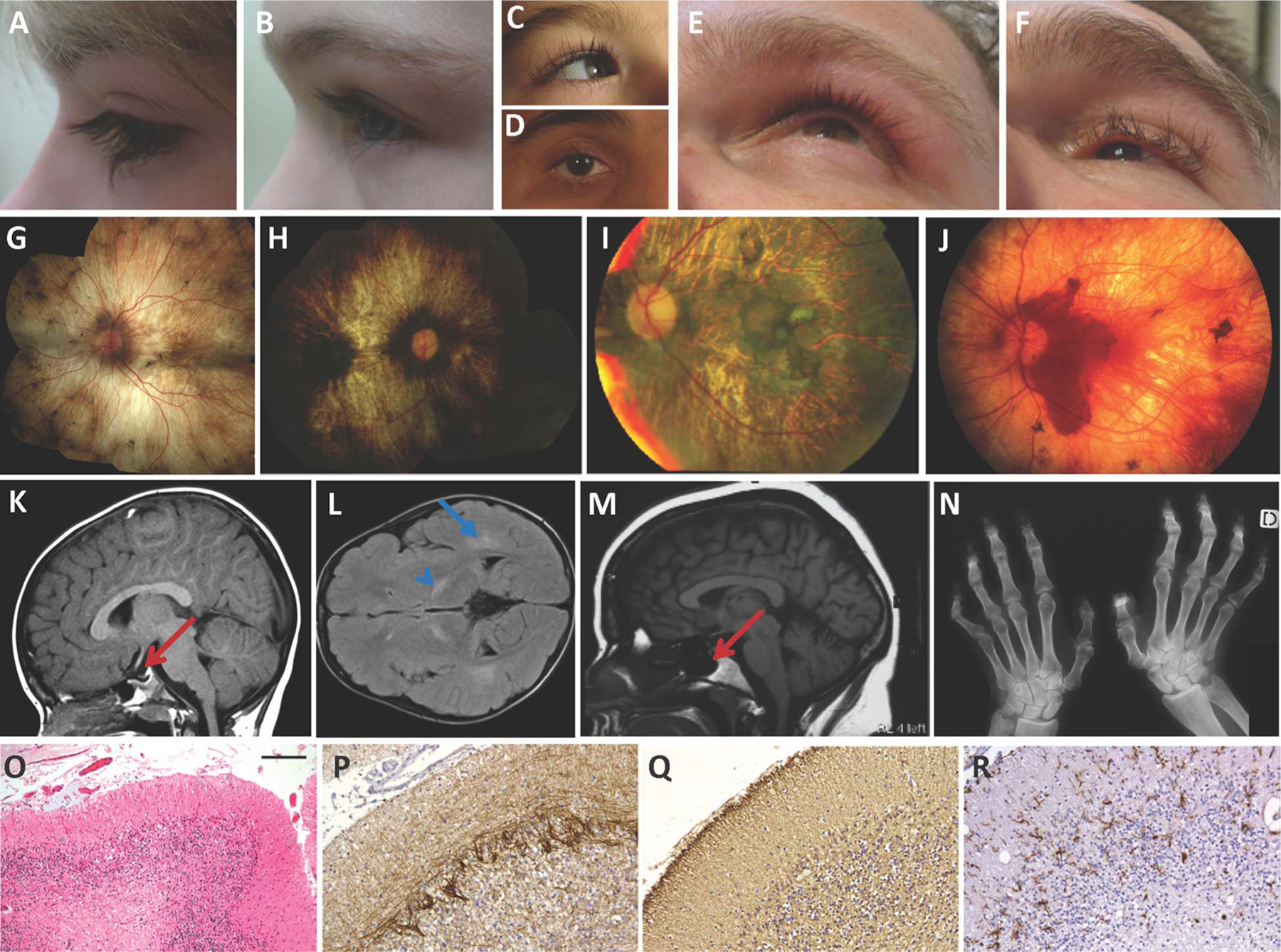
Background: Oliver-McFarlane syndrome is characterised by trichomegaly, congenital hypopituitarism and retinal degeneration with choroidal atrophy. Laurence-Moon syndrome presents similarly, though with progressive spinocerebellar ataxia and spastic paraplegia and without trichomegaly. Both recessively inherited disorders have no known genetic cause.
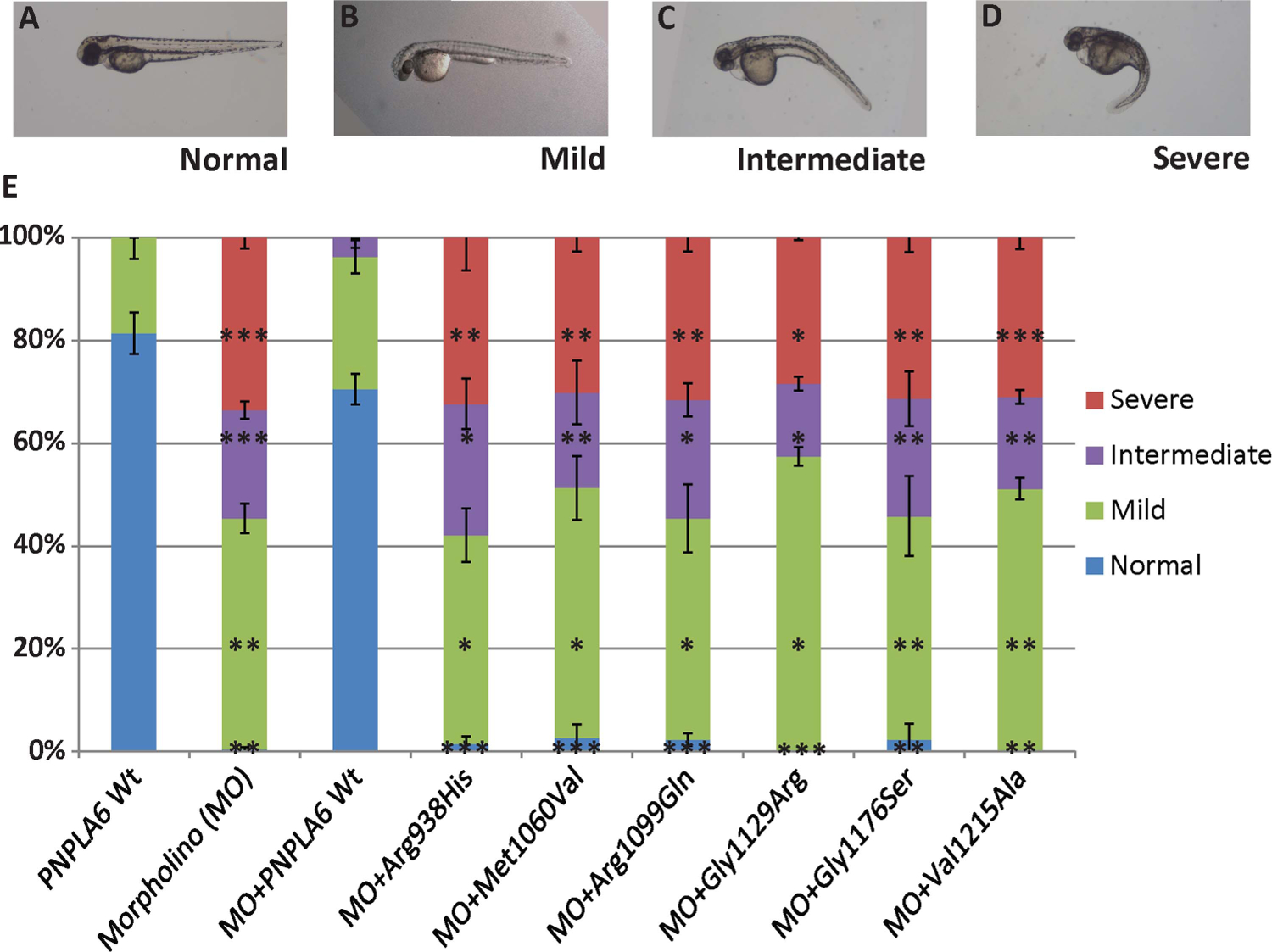
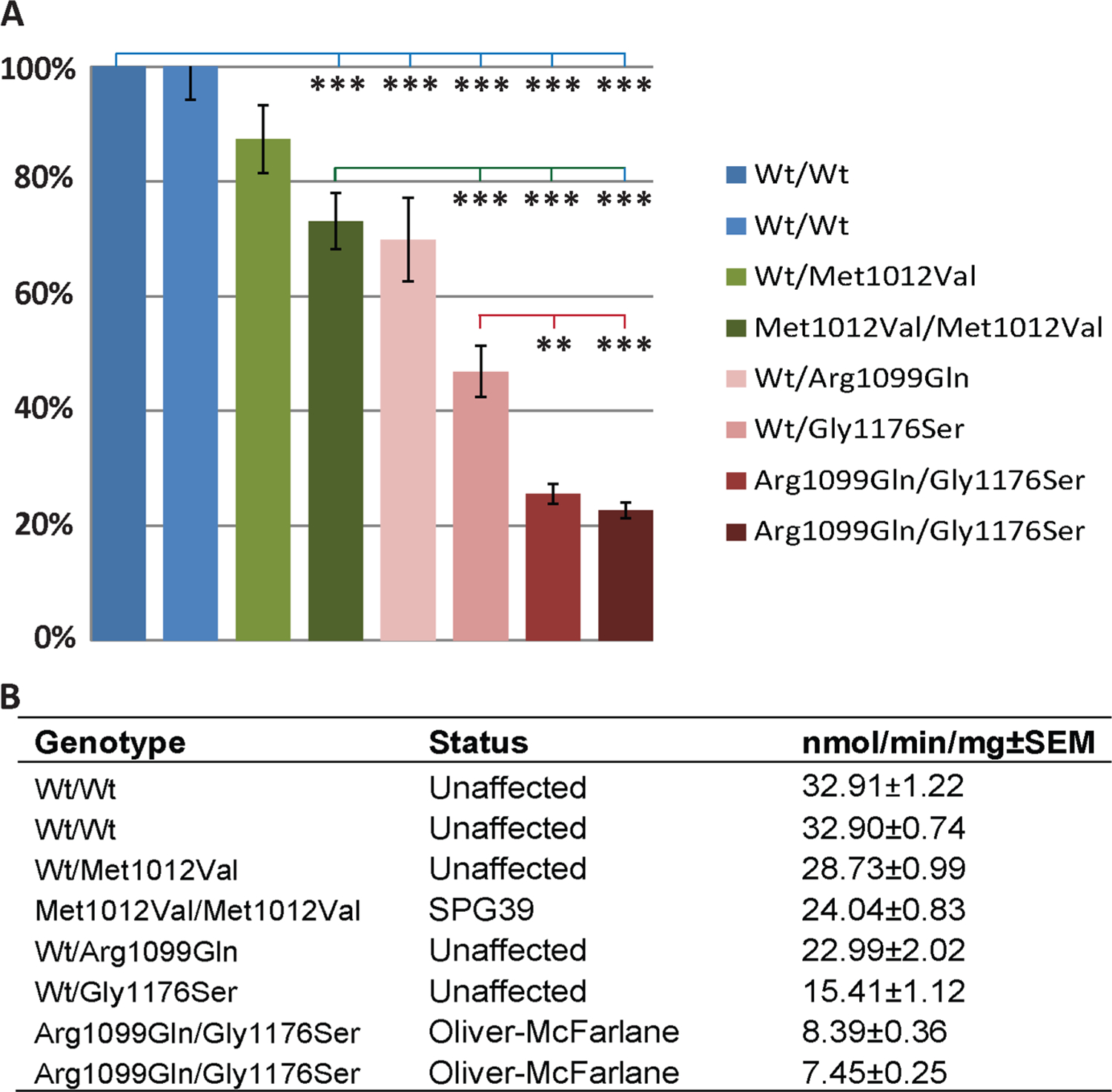
Methods: Whole-exome sequencing was performed to identify the genetic causes of these disorders. Mutations were functionally validated in zebrafish pnpla6 morphants. Embryonic expression was evaluated via in situ hybridisation in human embryonic sections. Human neurohistopathology was performed to characterise cerebellar degeneration. Enzymatic activities were measured in patient-derived fibroblast cell lines.
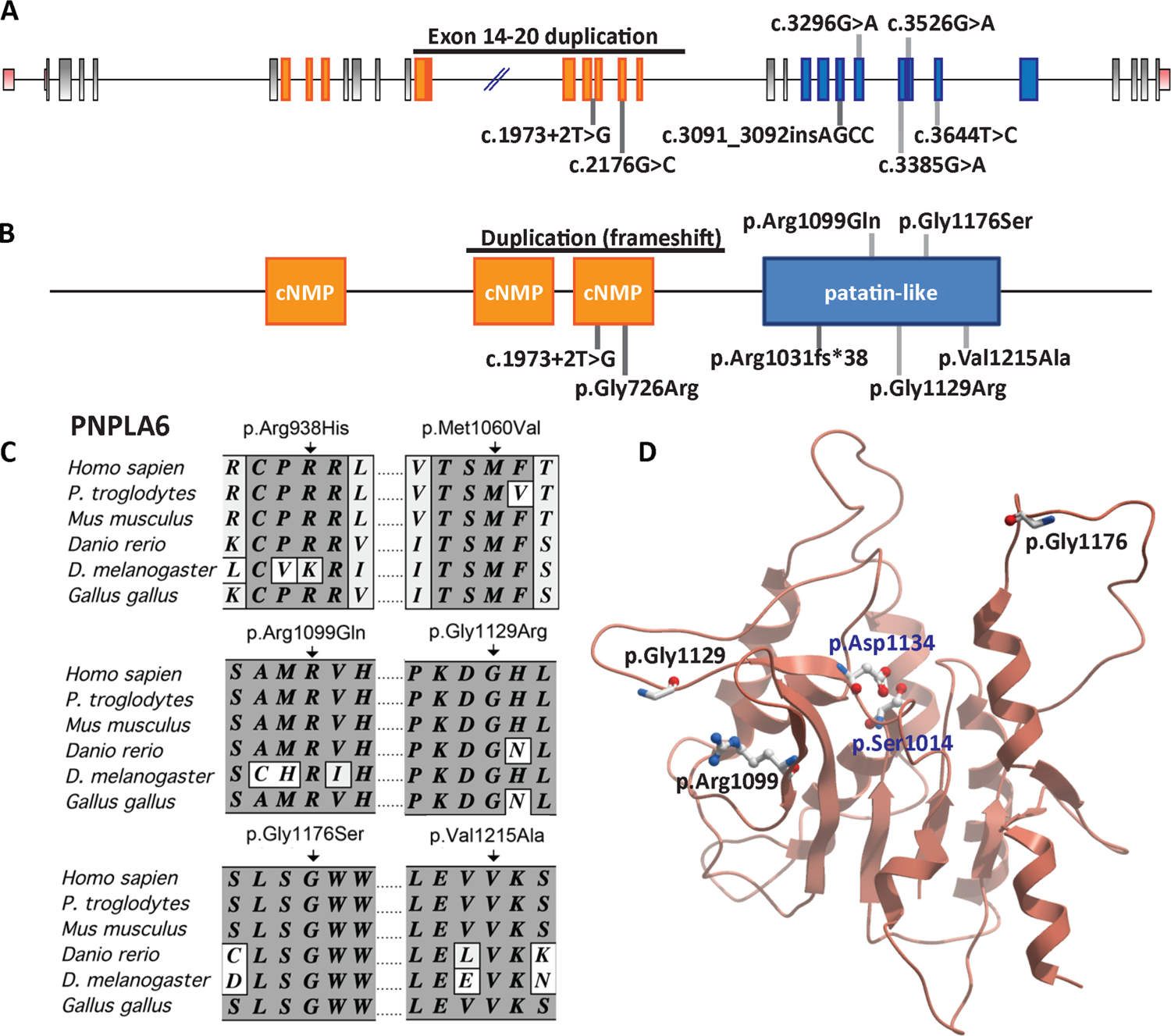
Results: Eight mutations in six families with Oliver-McFarlane or Laurence-Moon syndrome were identified in the PNPLA6 gene, which encodes neuropathy target esterase (NTE). PNPLA6 expression was found in the developing human eye, pituitary and brain. In zebrafish, the pnpla6 curly-tailed morphant phenotype was fully rescued by wild-type human PNPLA6 mRNA and not by mutation-harbouring mRNAs. NTE enzymatic activity was significantly reduced in fibroblast cells derived from individuals with Oliver-McFarlane syndrome. Intriguingly, adult brain histology from a patient with highly overlapping features of Oliver-McFarlane and Laurence-Moon syndromes revealed extensive cerebellar degeneration and atrophy.
Conclusions: Previously, PNPLA6 mutations have been associated with spastic paraplegia type 39, Gordon-Holmes syndrome and Boucher-Neuhäuser syndromes. Discovery of these additional PNPLA6-opathies further elucidates a spectrum of neurodevelopmental and neurodegenerative disorders associated with NTE impairment and suggests a unifying mechanism with diagnostic and prognostic importance.
Keywords: Dermatology; Developmental; Genetics; Neuro endocrinology; Ophthalmology.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://group.bmj.com/group/rights-licensing/permissions.
Conflict of interest statement
Figures

References
-
- Oliver GL, McFarlane DC. Congenital trichomegaly: with associated pigmentary degeneration of the retina, dwarfism, and mental retardation. Arch Ophthalmol 1965;74:169–71. - PubMed
-
- Chang TS, McFarlane DC, Oliver G, Willis NR. Congenital trichomegaly, pigmentary degeneration of the retina and growth retardation (Oliver-McFarlane syndrome): 28-year follow-up of the first reported case. Can J Ophthalmol 1993;28:191–3. - PubMed
-
- Corby DG, Lowe RS Jr, Haskins RC, Hebertson LM. Trichomegaly, pigmentary degeneration of the retina, and growth retardation. A new syndrome originating in utero. Am J Dis Child 1971;121:344–5. - PubMed
-
- Mathieu M, Goldfarb A, Berquin P, Boudailliez B, Labeille B, Piussan C. Trichomegaly, pigmentary degeneration of the retina and growth disturbances. a probable autosomal recessive disorder. Genet Couns 1991;2:115–8. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- MR/M000125/1/MRC_/Medical Research Council/United Kingdom
- R01 DC012564/DC/NIDCD NIH HHS/United States
- G1001253/MRC_/Medical Research Council/United Kingdom
- MC_PC_15004/MRC_/Medical Research Council/United Kingdom
- G108/638/MRC_/Medical Research Council/United Kingdom
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- G0802760/MRC_/Medical Research Council/United Kingdom
- 082557/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- UL1 TR000077/TR/NCATS NIH HHS/United States
- R01 DC011803/DC/NIDCD NIH HHS/United States
- G0700089/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials