

MED1 mediates androgen receptor splice variant induced gene expression in the absence of ligand
- PMID: 25481872
- PMCID: PMC4381595
- DOI: 10.18632/oncotarget.2672
MED1 mediates androgen receptor splice variant induced gene expression in the absence of ligand
Abstract
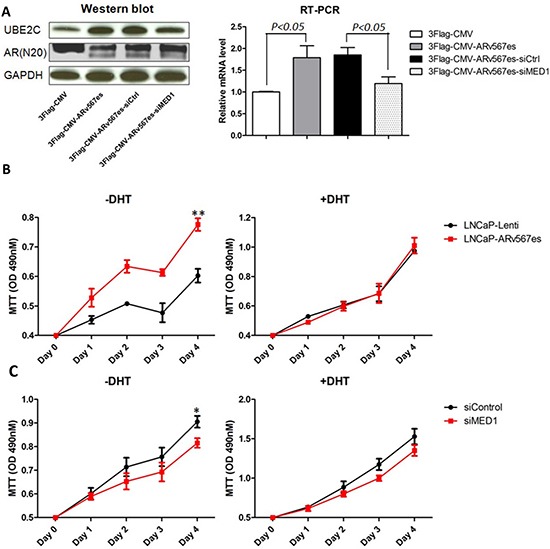
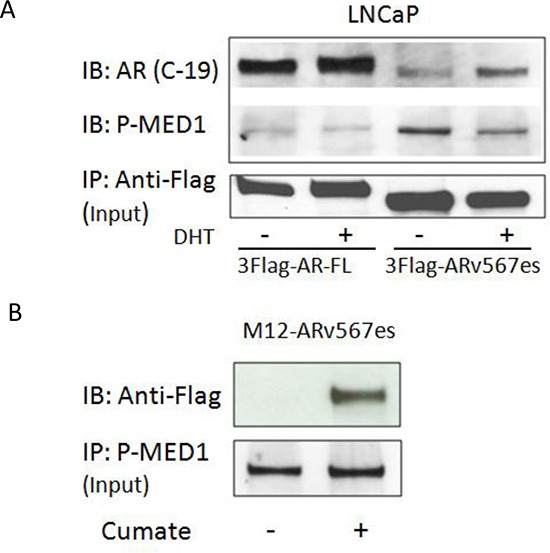
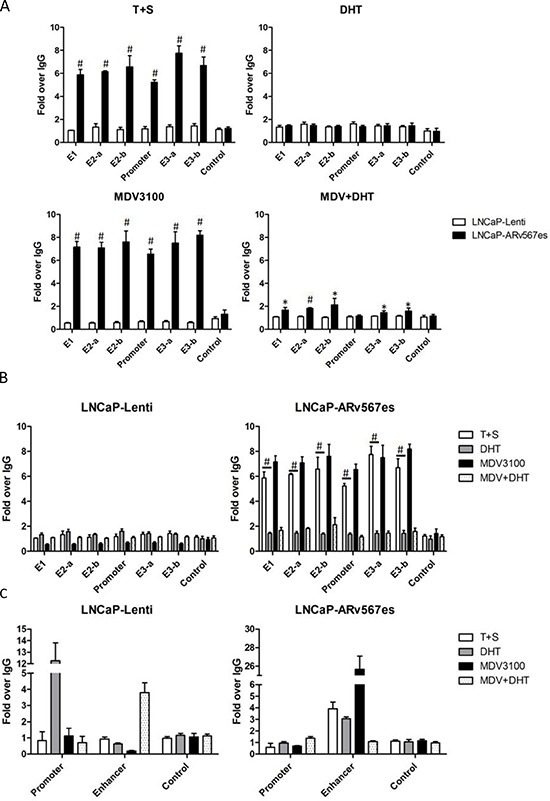
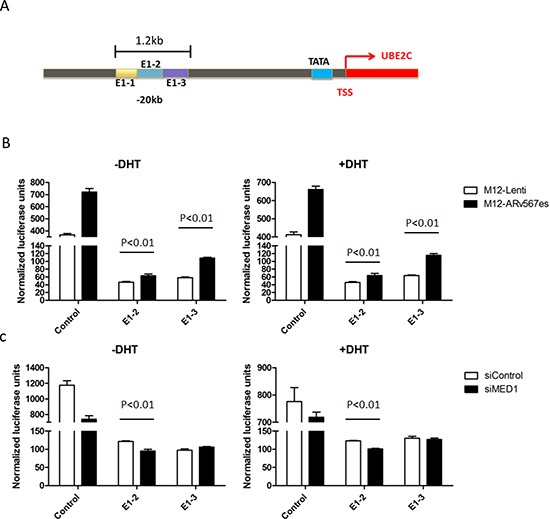
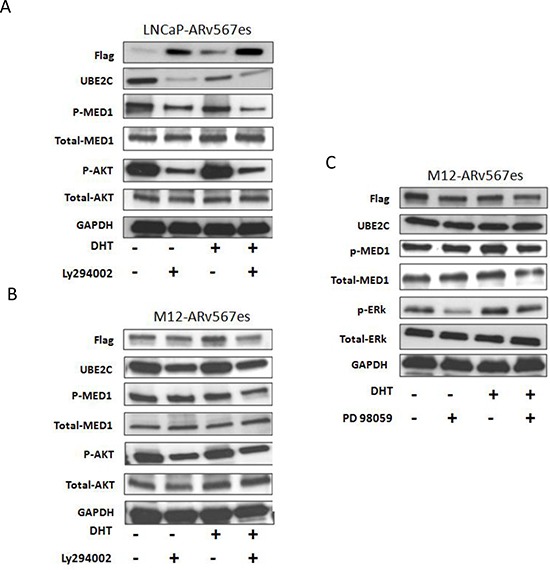
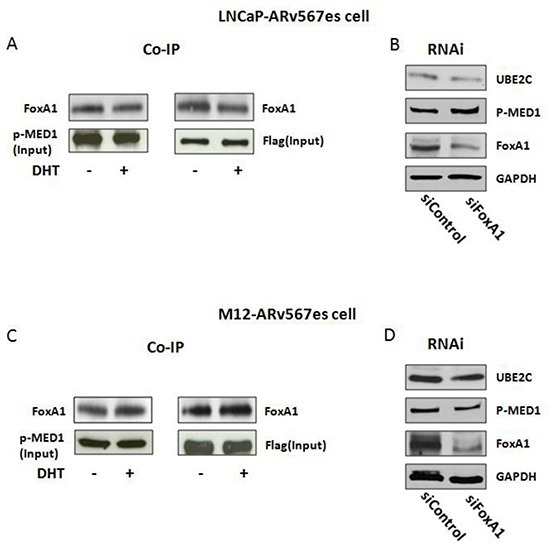
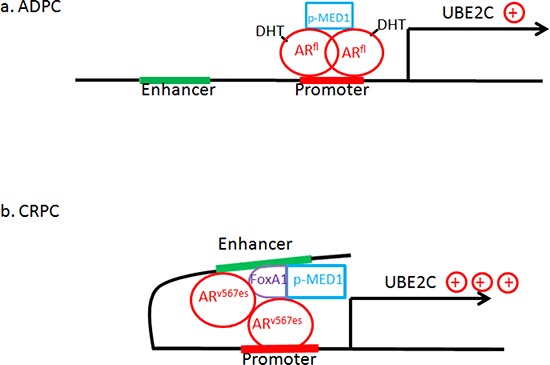
The appearance of constitutively active androgen receptor splice variants (AR-Vs) has been proposed as one of the causes of castration-resistant prostate cancer (CRPC). However, the underlying mechanism of AR-Vs in CRPC transcriptional regulation has not been defined. A distinct transcriptome enriched with cell cycle genes, e.g. UBE2C, has been associated with AR-Vs, which indicates the possibility of an altered transcriptional mechanism when compared to full-length wild-type AR (ARfl). Importantly, a recent study reported the critical role of p-MED1 in enhancing UBE2C expression through a locus looping pattern, which only occurs in CRPC but not in androgen-dependent prostate cancer (ADPC). To investigate the potential correlation between AR-V and MED1, in the present study we performed protein co-immunoprecipitation, chromatin immunoprecipitation, and cell proliferation assays and found that MED1 is necessary for ARv567es induced UBE2C up-regulation and subsequent prostate cancer cell growth. Furthermore, p-MED1 is bound to ARv567es independent of full-length AR; p-MED1 has higher recruitment to UBE2C promoter and enhancer regions in the presence of ARv567es. Our data indicate that p-MED1 serves as a key mediator in ARv567es induced gene expression and suggests a mechanism by which AR-Vs promote the development and progression of CRPC.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:3251–3255. - PubMed
-
- Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer research. 2008;68:6407–6415. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
