

Re-emergent human adenovirus genome type 7d caused an acute respiratory disease outbreak in Southern China after a twenty-one year absence
- PMID: 25482188
- PMCID: PMC4258649
- DOI: 10.1038/srep07365
Re-emergent human adenovirus genome type 7d caused an acute respiratory disease outbreak in Southern China after a twenty-one year absence
Abstract
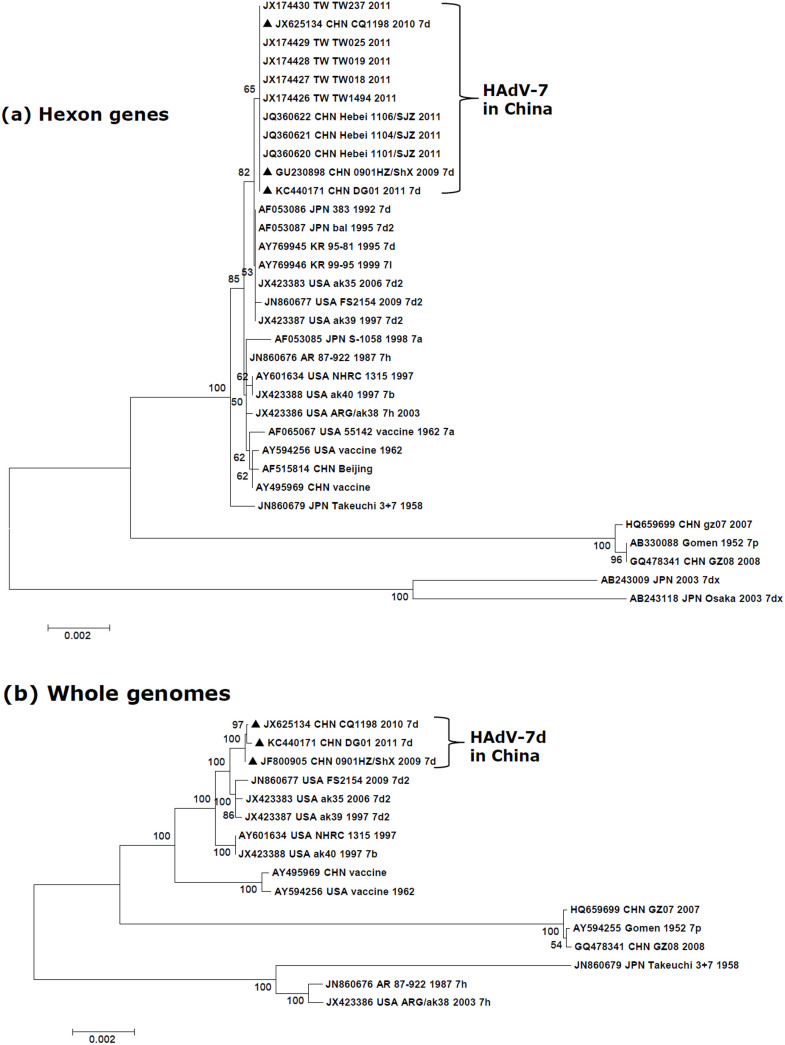
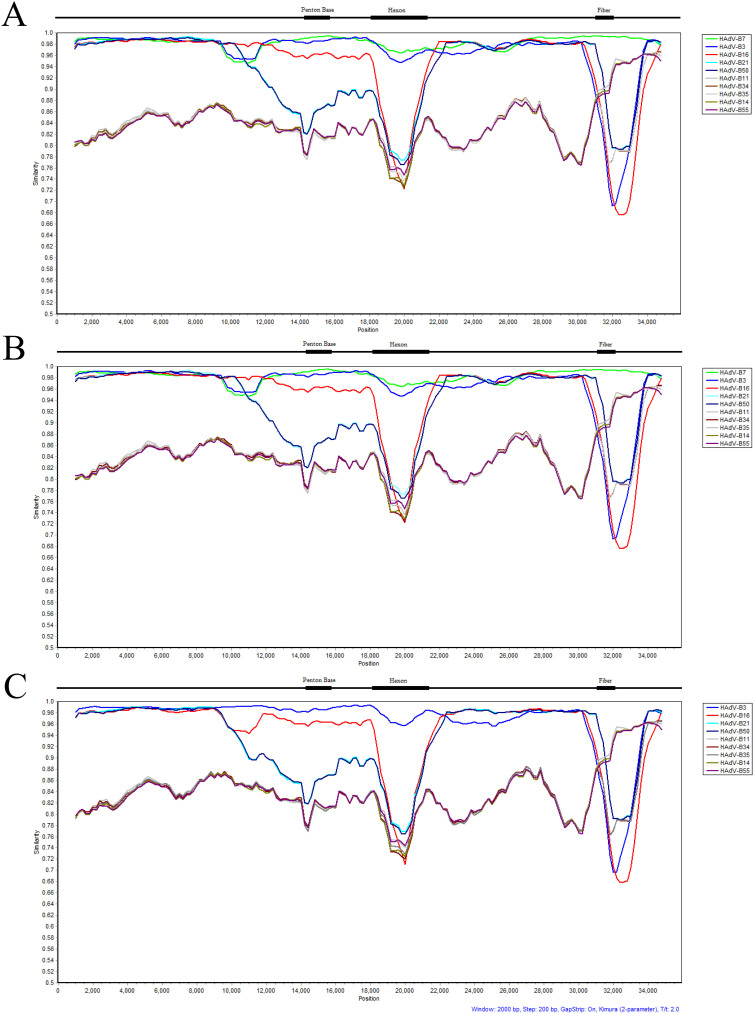
Human adenoviruses (HAdVs) are highly contagious pathogens causing acute respiratory disease (ARD), among other illnesses. Of the ARD genotypes, HAdV-7 presents with more severe morbidity and higher mortality than the others. We report the isolation and identification of a genome type HAdV-7d (DG01_2011) from a recent outbreak in Southern China. Genome sequencing, phylogenetic analysis, and restriction endonuclease analysis (REA) comparisons with past pathogens indicate HAdV-7d has re-emerged in Southern China after an absence of twenty-one years. Recombination analysis reveals this genome differs from the 1950s-era prototype and vaccine strains by a lateral gene transfer, substituting the coding region for the L1 52/55 kDa DNA packaging protein from HAdV-16. DG01_2011 descends from both a strain circulating in Southwestern China (2010) and a strain from Shaanxi causing a fatality and outbreak (Northwestern China; 2009). Due to the higher morbidity and mortality rates associated with HAdV-7, the surveillance, identification, and characterization of these strains in population-dense China by REA and/or whole genome sequencing are strongly indicated. With these accurate identifications of specific HAdV types and an epidemiological database of regional HAdV pathogens, along with the HAdV genome stability noted across time and space, the development, availability, and deployment of appropriate vaccines are needed.
Figures

References
-
- Hierholzer J. C. in Diagnostic Procedures for Viral, Rickettsial, and Chlamydial Infections (eds Lennette, E. H., Lennette, D. A. & Lennette, E. T.) 169–188 (American Public Health Association, 1995).
-
- Seto D., Jones M. S., Dyer D. W. & Chodosh J. Characterizing, typing, and naming human adenovirus type 55 in the era of whole genome data. J. Clin. Virol. 58, 741–742 (2013). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
