

Increasing cisplatin sensitivity by schedule-dependent inhibition of AKT and Chk1
- PMID: 25482935
- PMCID: PMC4623033
- DOI: 10.4161/15384047.2014.961876
Increasing cisplatin sensitivity by schedule-dependent inhibition of AKT and Chk1
Abstract
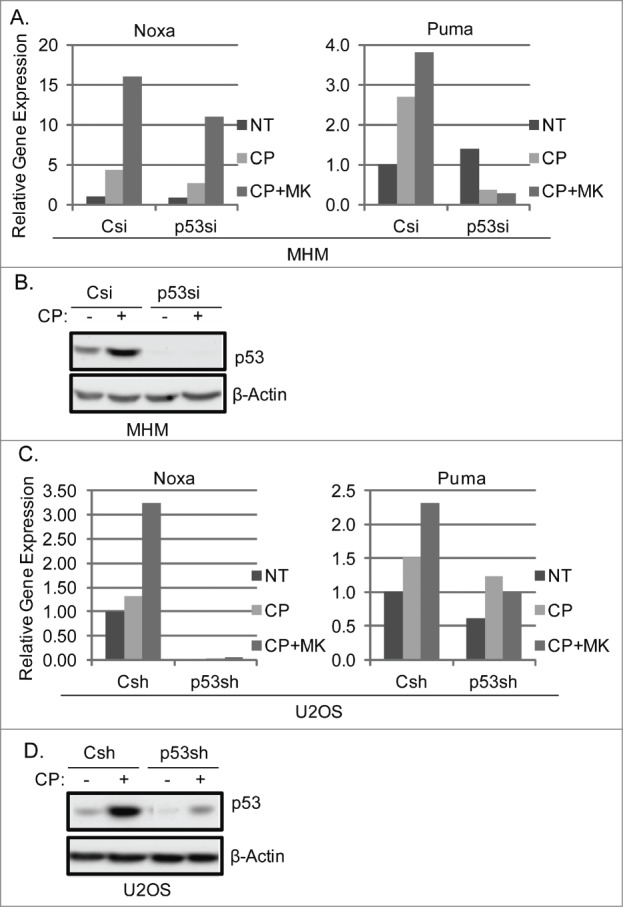
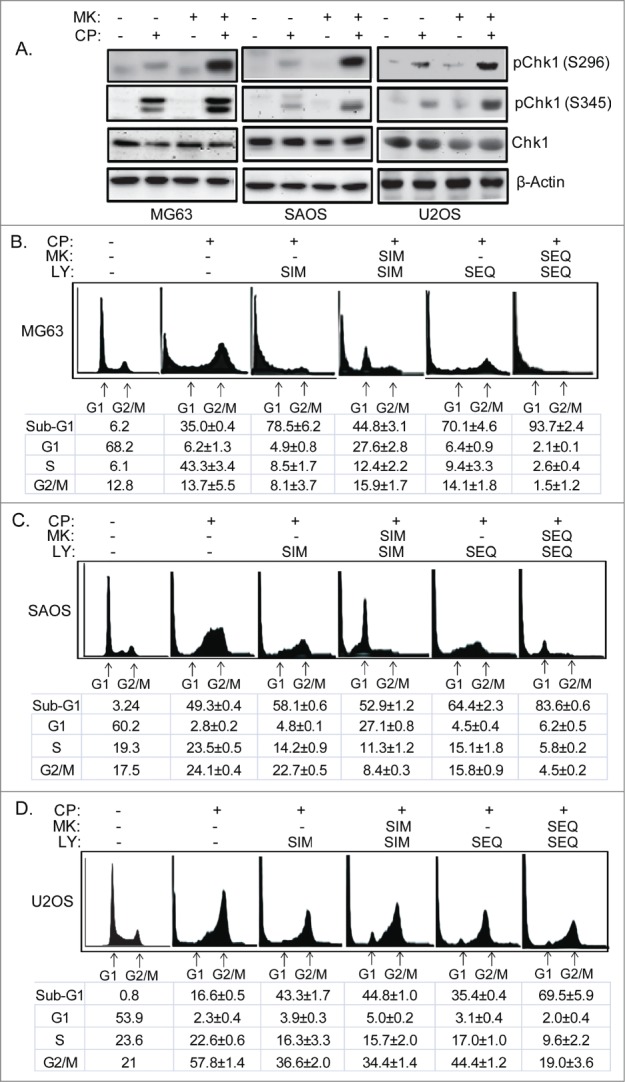
The effectiveness of DNA damaging chemotherapy drugs can be limited by activation of survival signaling pathways and cell cycle checkpoints that allow DNA repair. Targeting survival pathways and inhibiting cell cycle checkpoints may increase chemotherapy-induced cancer cell killing. AKT and Chk1 are survival and cell cycle checkpoint kinases, respectively, that can be activated by DNA damage. Cisplatin (CP) is a standard chemotherapy agent for osteosarcoma (OS). CP induced apoptosis to varying extents and activated AKT and Chk1 in multiple p53 wild-type and p53-null OS cell lines. A Chk1 inhibitor increased CP-induced apoptosis in all OS cell lines regardless of p53 status. In contrast, an AKT inhibitor increased CP-induced apoptosis only in p53 wild-type OS cells, but not p53 nulll cells. The increased apoptosis in p53 wild-type cells was coincident with decreased p53 protein levels, but increased expression of p53-responsive apoptotic genes Noxa and PUMA. Further studies revealed the inability of AKT inhibitor to CP-sensitize p53-null OS cells resulted from 2 things: 1) AKT inhibition stabilized/maintained p27 levels in CP-treated cells, which then mediated a protective G1-phase cell cycle arrest, 2) AKT inhibition increased the levels of activated Chk1. Finally, schedule dependent inhibition of AKT and Chk1 evaded the protective G1 arrest mediated by p27 and maximized CP-induced OS cell killing. These data demonstrate AKT and Chk1 activation promote survival in CP-treated OS cells, and that strategic, scheduled targeting of AKT and Chk1 can maximize OS cell killing by CP.
Keywords: AKT; CDC2, cell division cycle kinase 2; Chk1; Chk1, Checkpoint kinase 1; apoptosis; cisplatin; mTORC1 and mTORC2, mammalian target of rapamycin complex 1 and 2; osteosarcoma; p53.
Figures









References
-
- Eastman A. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem 2004; 91:223-31; PMID:14743382; http://dx.doi.org/ 10.1002/jcb.10699 - DOI - PubMed
-
- Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell 2012; 23:1-6; PMID:22210845; http://dx.doi.org/ 10.1091/mbc.E10-04-0335 - DOI - PMC - PubMed
-
- Lu C, Fu W, Zhao D, Mattson MP. The DNA damaging agent etoposide activates a cell survival pathway involving alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors and mitogen-activated protein kinases in hippocampal neurons. J Neurosci Res 2002; 70:671-9; PMID:12424735; http://dx.doi.org/ 10.1002/jnr.10413 - DOI - PubMed
-
- Wang X, McCullough KD, Franke TF, Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem 2000; 275:14624-31; PMID:10799549; http://dx.doi.org/ 10.1074/jbc.275.19.14624 - DOI - PubMed
-
- Li HF, Kim JS, Waldman T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiation Oncol 2009; 4:43; http://dx.doi.org/ 10.1186/1748-717X-4-43 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous