

Regulation of dimethyl-fumarate toxicity by proteasome inhibitors
- PMID: 25482938
- PMCID: PMC4623310
- DOI: 10.4161/15384047.2014.967992
Regulation of dimethyl-fumarate toxicity by proteasome inhibitors
Abstract
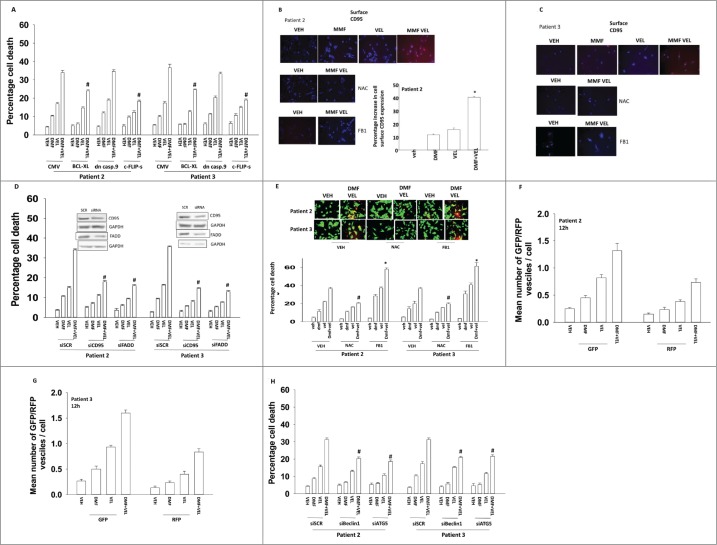
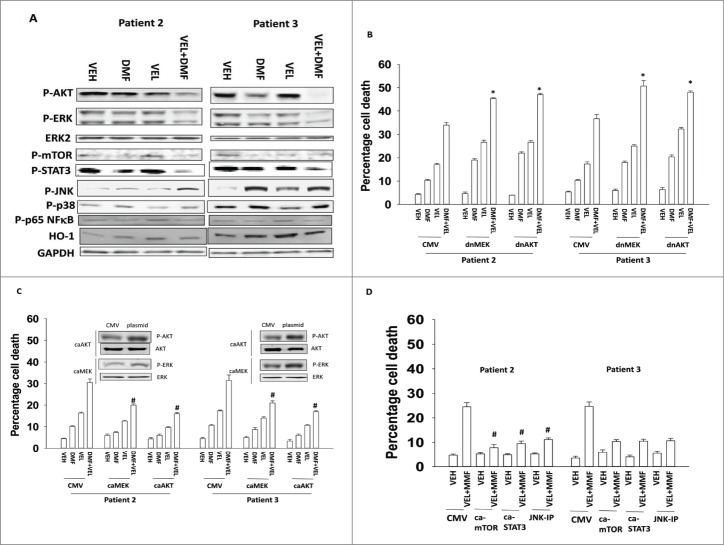
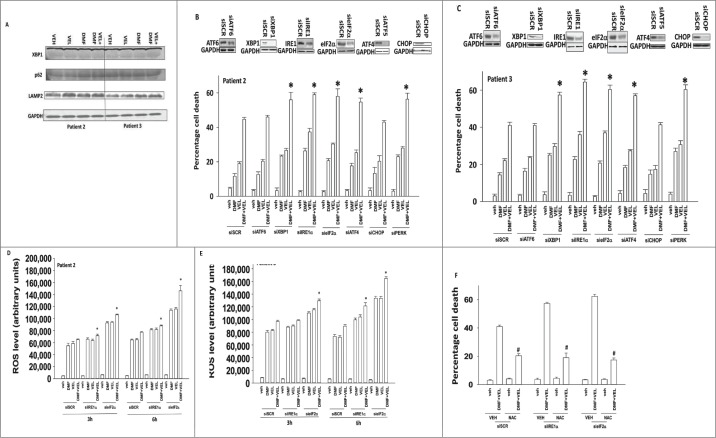
The present studies examined the biology of the multiple sclerosis drug dimethyl-fumarate (DMF) or its in vivo breakdown product and active metabolite mono-methyl-fumarate (MMF), alone or in combination with proteasome inhibitors, in primary human glioblastoma (GBM) cells. MMF enhanced velcade and carfilzomib toxicity in multiple primary GBM isolates. Similar data were obtained in breast and colon cancer cells. MMF reduced the invasiveness of GBM cells, and enhanced the toxicity of ionizing radiation and temozolomide. MMF killed freshly isolated activated microglia which was associated with reduced IL-6, TGFβ and TNFα production. The combination of MMF and the multiple sclerosis drug Gilenya further reduced both GBM and activated microglia viability and cytokine production. Over-expression of c-FLIP-s or BCL(-)XL protected GBM cells from MMF and velcade toxicity. MMF and velcade increased plasma membrane localization of CD95, and knock down of CD95 or FADD blocked the drug interaction. The drug combination inactivated AKT, ERK1/2 and mTOR. Molecular inhibition of AKT/ERK/mTOR signaling enhanced drug combination toxicity whereas molecular activation of these pathways suppressed killing. MMF and velcade increased the levels of autophagosomes and autolysosomes and knock down of ATG5 or Beclin1 protected cells. Inhibition of the eIF2α/ATF4 arm or the IRE1α/XBP1 arm of the ER stress response enhanced drug combination lethality. This was associated with greater production of reactive oxygen species and quenching of ROS suppressed cell killing.
Keywords: DMF, dimethyl-fumarate; EGF, epidermal growth factor; ERK, extracellular regulated kinase; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen activated protein kinase; MEK, mitogen activated extracellular regulated kinase; MMF, monomethyl-fumarate; P, phospho-; PARP, poly ADP ribosyl polymerase; PI3K, phosphatidyl inositol 3 kinase; PTEN, Phosphatase and tensin homolog; R, receptor; WT, wild type; ca, constitutively active; dn, dominant negative; −/−, null / gene deleted.
Figures
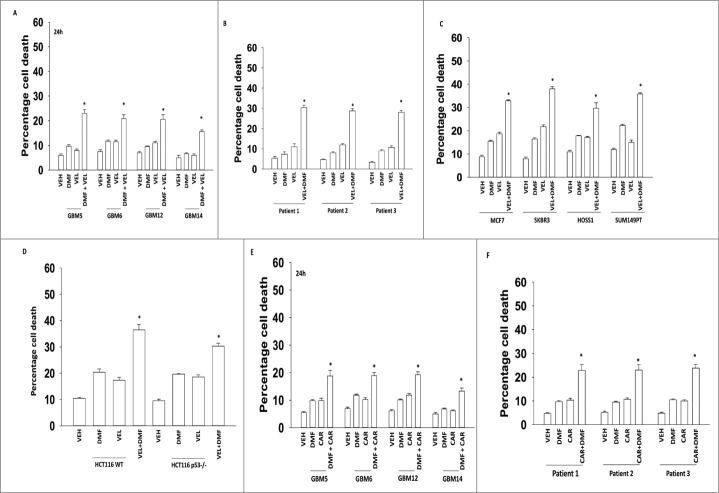
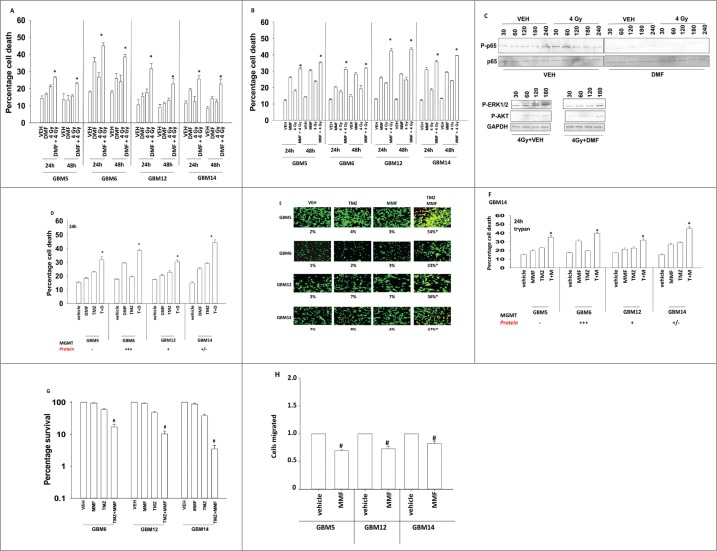
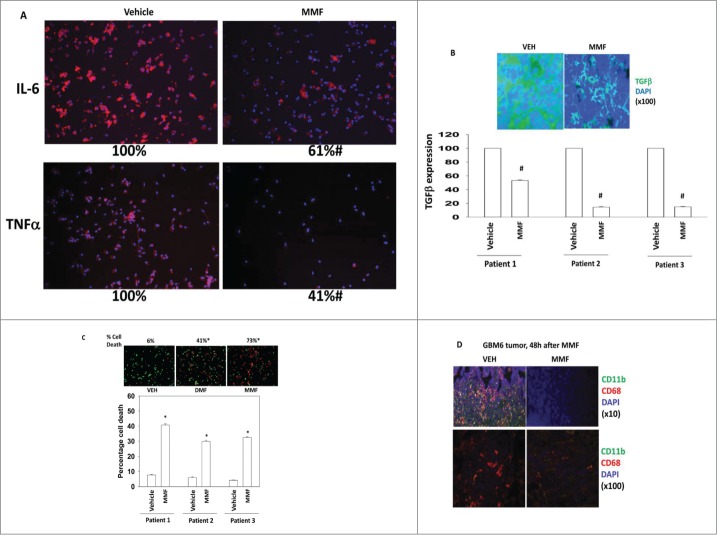
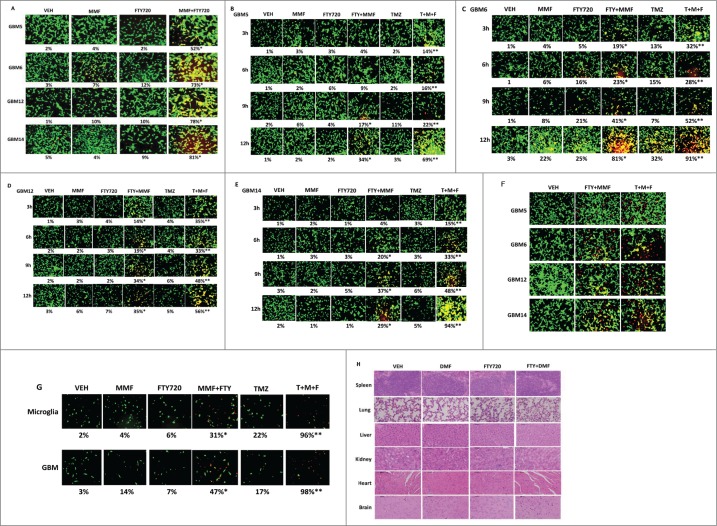
Similar articles
-
Ruxolitinib synergizes with DMF to kill via BIM+BAD-induced mitochondrial dysfunction and via reduced SOD2/TRX expression and ROS.Oncotarget. 2016 Apr 5;7(14):17290-300. doi: 10.18632/oncotarget.8039. Oncotarget. 2016. PMID: 26981780 Free PMC article.
-
Fingolimod Augments Monomethylfumarate Killing of GBM Cells.Front Oncol. 2020 Jan 28;10:22. doi: 10.3389/fonc.2020.00022. eCollection 2020. Front Oncol. 2020. PMID: 32047722 Free PMC article.
-
Differential regulation of autophagy and cell viability by ceramide species.Cancer Biol Ther. 2015;16(5):733-42. doi: 10.1080/15384047.2015.1026509. Cancer Biol Ther. 2015. PMID: 25803131 Free PMC article.
-
New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia.Cell Signal. 2014 Jan;26(1):149-61. doi: 10.1016/j.cellsig.2013.09.021. Epub 2013 Oct 16. Cell Signal. 2014. PMID: 24140475 Review.
-
Dimethyl fumarate downregulates the immune response through the HCA2/GPR109A pathway: Implications for the treatment of multiple sclerosis.Mult Scler Relat Disord. 2018 Jul;23:46-50. doi: 10.1016/j.msard.2018.04.016. Epub 2018 Apr 25. Mult Scler Relat Disord. 2018. PMID: 29763776 Review.
Cited by
-
Molecular Effects of FDA-Approved Multiple Sclerosis Drugs on Glial Cells and Neurons of the Central Nervous System.Int J Mol Sci. 2020 Jun 13;21(12):4229. doi: 10.3390/ijms21124229. Int J Mol Sci. 2020. PMID: 32545828 Free PMC article. Review.
-
Phase I trial of dimethyl fumarate, temozolomide, and radiation therapy in glioblastoma.Neurooncol Adv. 2020 Jan 24;2(1):vdz052. doi: 10.1093/noajnl/vdz052. eCollection 2020 Jan-Dec. Neurooncol Adv. 2020. PMID: 32642720 Free PMC article.
-
Ruxolitinib synergizes with DMF to kill via BIM+BAD-induced mitochondrial dysfunction and via reduced SOD2/TRX expression and ROS.Oncotarget. 2016 Apr 5;7(14):17290-300. doi: 10.18632/oncotarget.8039. Oncotarget. 2016. PMID: 26981780 Free PMC article.
-
Roles of Glutathione and AP-1 in the Enhancement of Vitamin D-Induced Differentiation by Activators of the Nrf2 Signaling Pathway in Acute Myeloid Leukemia Cells.Int J Mol Sci. 2024 Feb 14;25(4):2284. doi: 10.3390/ijms25042284. Int J Mol Sci. 2024. PMID: 38396960 Free PMC article.
-
Therapeutic Targeting of TAZ and YAP by Dimethyl Fumarate in Systemic Sclerosis Fibrosis.J Invest Dermatol. 2018 Jan;138(1):78-88. doi: 10.1016/j.jid.2017.08.024. Epub 2017 Sep 1. J Invest Dermatol. 2018. PMID: 28870693 Free PMC article.
References
-
- Field KM, Rosenthal MA, Yilmaz M, Tacey M, Drummond K. Comparison between poor and long-term survivors with glioblastoma: review of an Australian data set. Asia Pac J Clin Oncol 2013; 10:153-61 [Epub ahead of print]; PMID:23701649 - PubMed
-
- Juratli TA, Schackert G, Krex D. Current status of local therapy in malignant gliomas–A clinical review of three selected approaches. Pharmacol Ther 2013; 139:341-58; PMID:23694764; http://dx.doi.org/10.1016/j.pharmthera.2013.05.003 - DOI - PubMed
-
- Phillips JT, Fox RJ. BG-12 in multiple sclerosis. Semin Neurol 2013; 33:56-65; PMID:23709213; http://dx.doi.org/10.1055/s-0033-1343796 - DOI - PubMed
-
- Linker RA, Lee DH, Ryan S, Van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, et al. . Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011; 134:678-692; PMID:21354971; http://dx.doi.org/10.1093/brain/awq386 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous