

Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents
- PMID: 25483091
- PMCID: PMC4615027
- DOI: 10.4161/15384101.2014.960729
Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents
Abstract
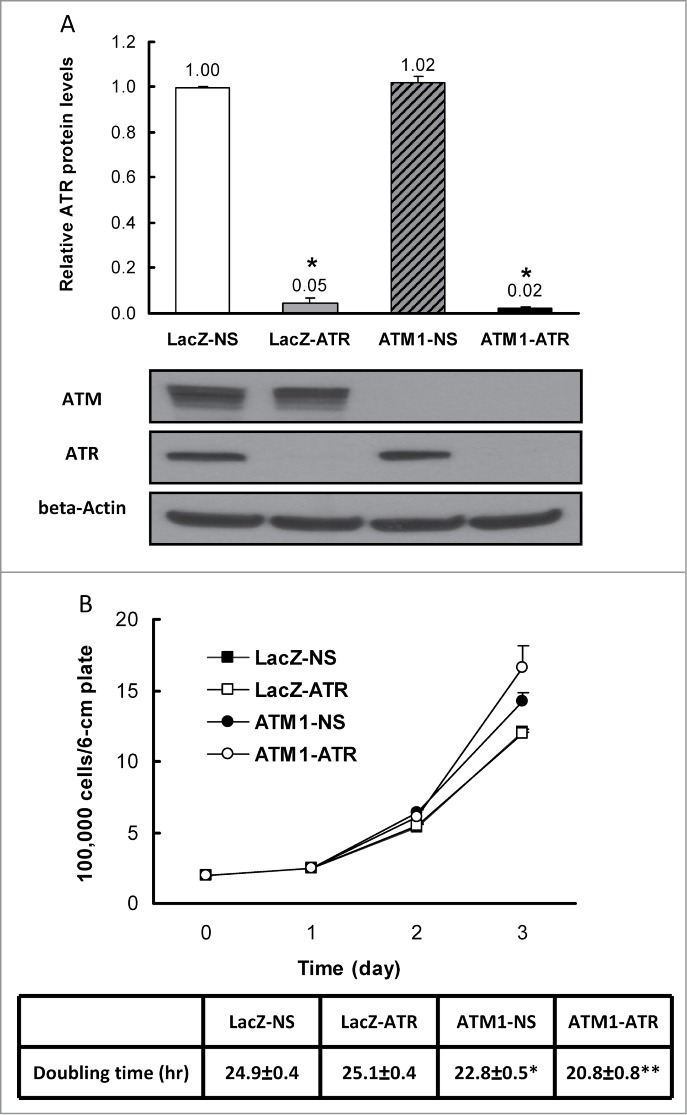
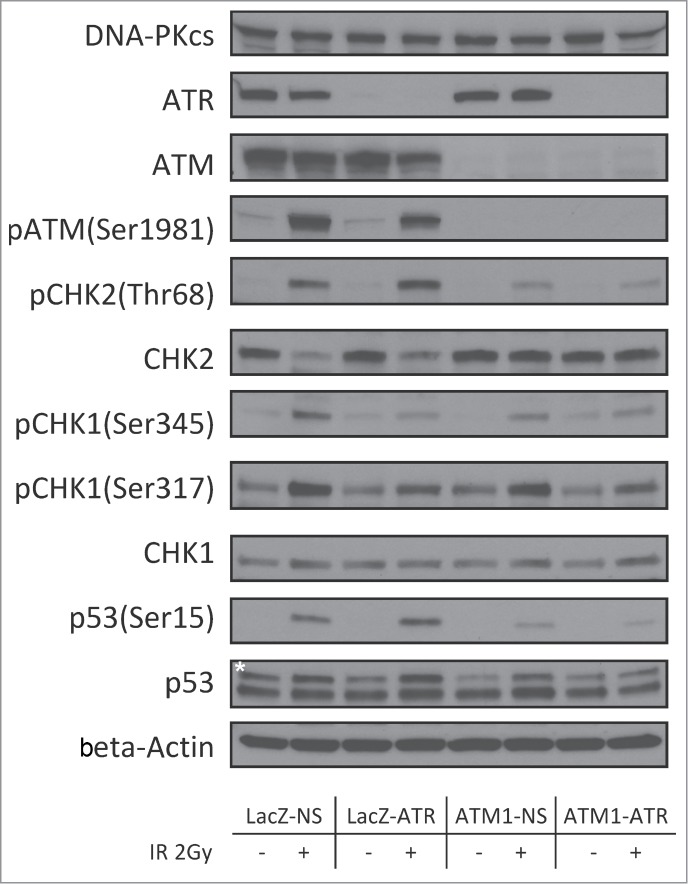
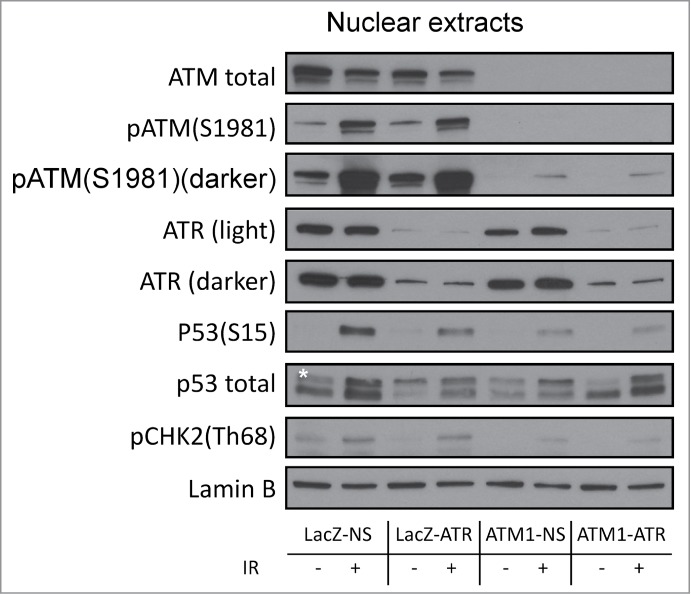
DNA damage response (DDR) to double strand breaks is coordinated by 3 phosphatidylinositol 3-kinase-related kinase (PIKK) family members: the ataxia-telangiectasia mutated kinase (ATM), the ATM and Rad3-related (ATR) kinase and the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). ATM and ATR are central players in activating cell cycle checkpoints and function as an active barrier against genome instability and tumorigenesis in replicating cells. Loss of ATM function is frequently reported in various types of tumors, thus placing more reliance on ATR for checkpoint arrest and cell survival following DNA damage. To investigate the role of ATR in the G2/M checkpoint regulation in response to ionizing radiation (IR), particularly when ATM is deficient, cell lines deficient of ATM, ATR, or both were generated using a doxycycline-inducible lentiviral system. Our data suggests that while depletion of ATR or ATM alone in wild-type human mammary epithelial cell cultures (HME-CCs) has little effect on radiosensitivity or IR-induced G2/M checkpoint arrest, depletion of ATR in ATM-deficient cells causes synthetic lethality following IR, which correlates with severe G2/M checkpoint attenuation. ATR depletion also inhibits IR-induced autophagy, regardless of the ATM status, and enhances IR-induced apoptosis particularly when ATM is deficient. Collectively, our results clearly demonstrate that ATR function is required for the IR-induced G2/M checkpoint activation and subsequent survival of cells with ATM deficiency. The synthetic lethal interaction between ATM and ATR in response to IR supports ATR as a therapeutic target for improved anti-cancer regimens, especially in tumors with a dysfunctional ATM pathway.
Keywords: ATM and Rad3-related (ATR); ATM, the ataxia-telangiectasia mutated kinase; ATP, adenosine triphosphate; ATR, the ATM and Rad3-related; CHK1, the checkpoint kinase 1; CHK2, the checkpoint kinase 2; DAPI, 4′,6-diamidino-2-phenylindole; DDR, DNA damage response; DNA damage response; DNA-PKcs, the catalytic subunit of the DNA-dependent protein kinase; DSBs, double strand breaks; G2/M checkpoint; HME-CCs, human mammary epithelial cell cultures; IR, ionizing radiation; RMI, relative mitotic index; SSBs, single strand breaks; WT, Wild-type; ionizing radiation; synthetic lethality.
Figures



References
-
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GCM, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37-U13; PMID:16327781; http://dx.doi.org/10.1038/ncb1337 - DOI - PubMed
-
- Zou L, Shiotani B. Single-Stranded DNA Orchestrates an ATM-to-ATR Switch at DNA Breaks. Mol Cell 2009; 33:547-58; PMID:19285939; http://dx.doi.org/10.1016/j.molcel.2009.01.024 - DOI - PMC - PubMed
-
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao ZM, Solimini N, Lerenthal Y, et al. . ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160-6; PMID:17525332; http://dx.doi.org/10.1126/science.1140321 - DOI - PubMed
-
- Smith J, Tho LM, Xu NH, Gillespie DA. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. Advances in Cancer Research, Vol 108 San Diego: Elsevier Academic Press Inc, 2010:73-112. - PubMed
-
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17:615-28; PMID:12629044; http://dx.doi.org/10.1101/gad.1067403 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous