

Aspartate β-Hydroxylase expression promotes a malignant pancreatic cellular phenotype
- PMID: 25483102
- PMCID: PMC4359229
- DOI: 10.18632/oncotarget.2840
Aspartate β-Hydroxylase expression promotes a malignant pancreatic cellular phenotype
Erratum in
-
Correction: Aspartate β-hydroxylase expression promotes a malignant pancreatic cellular phenotype.Oncotarget. 2019 Nov 12;10(61):6644-6646. doi: 10.18632/oncotarget.27315. eCollection 2019 Nov 12. Oncotarget. 2019. PMID: 31762945 Free PMC article.
Abstract
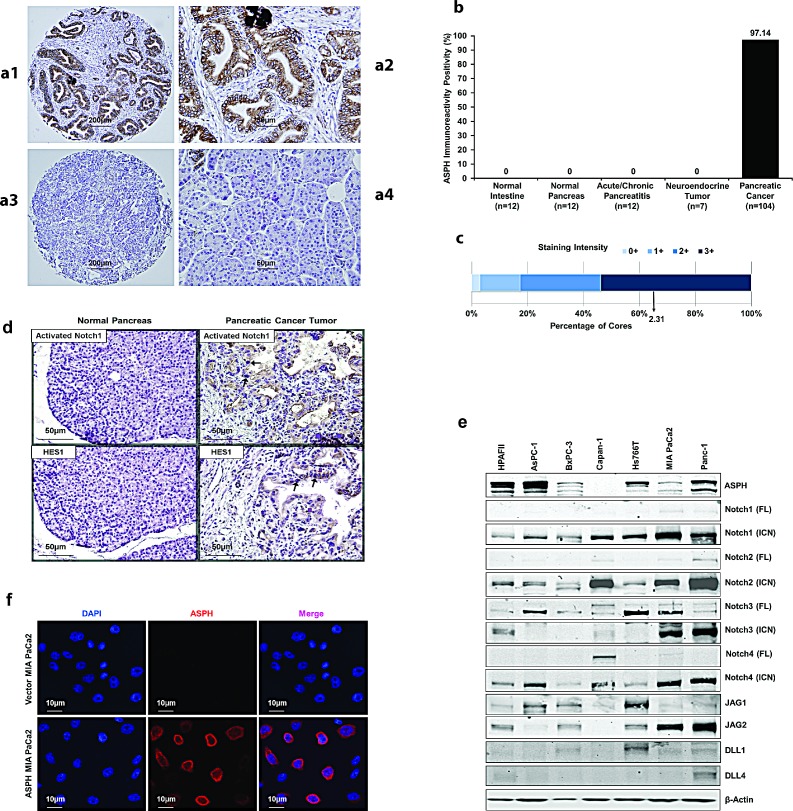
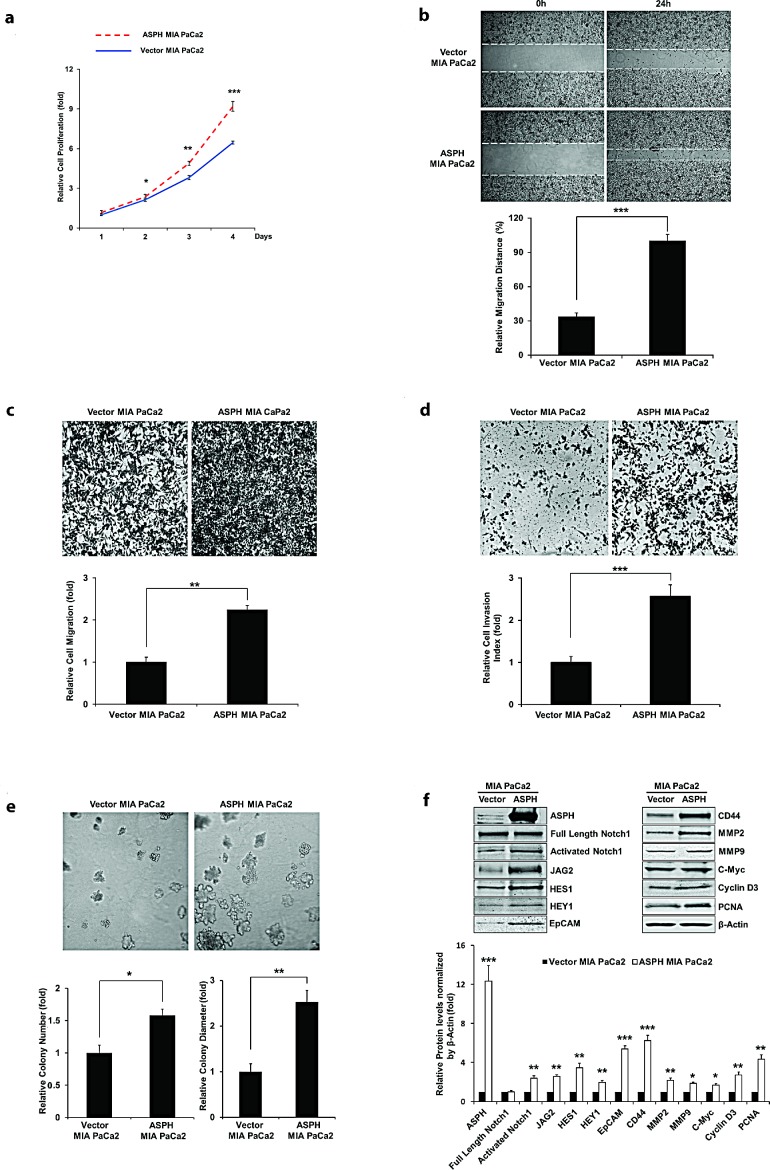
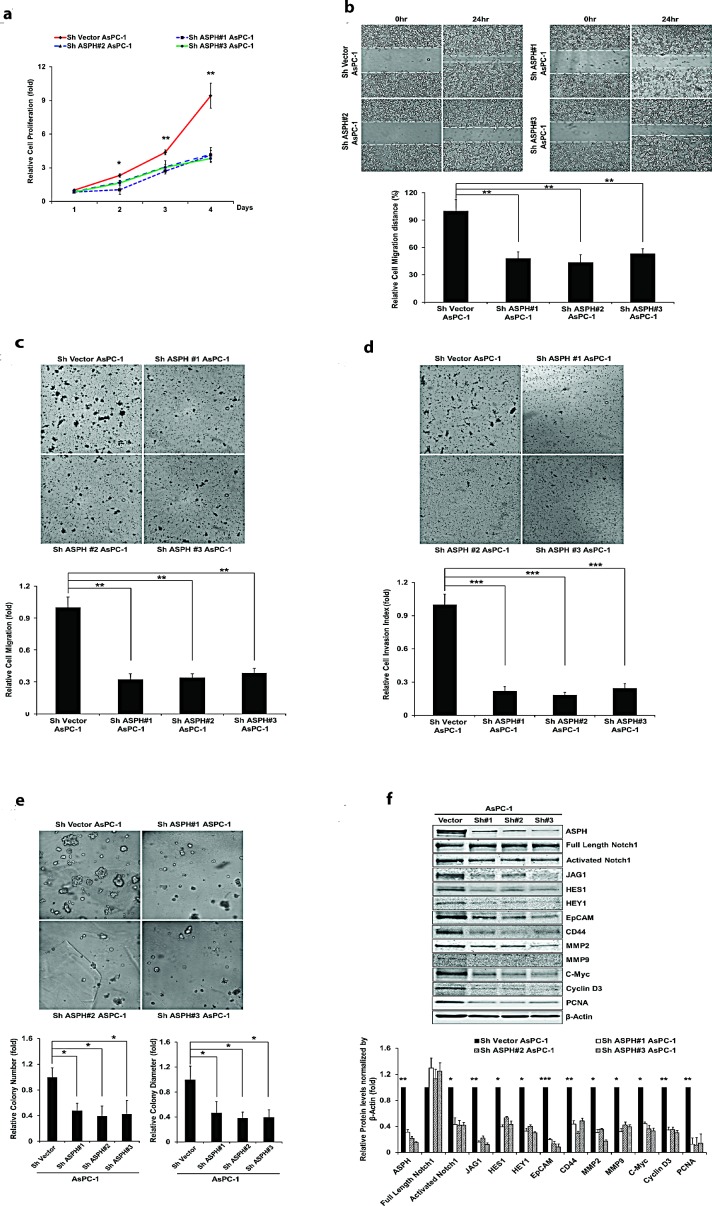
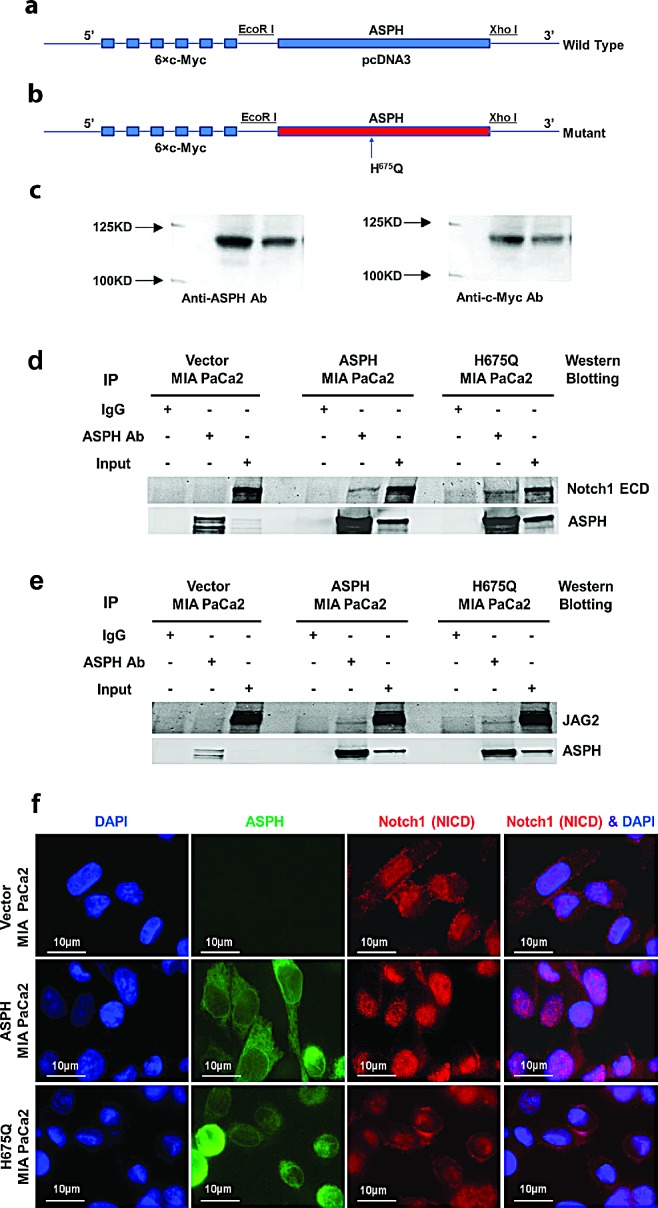
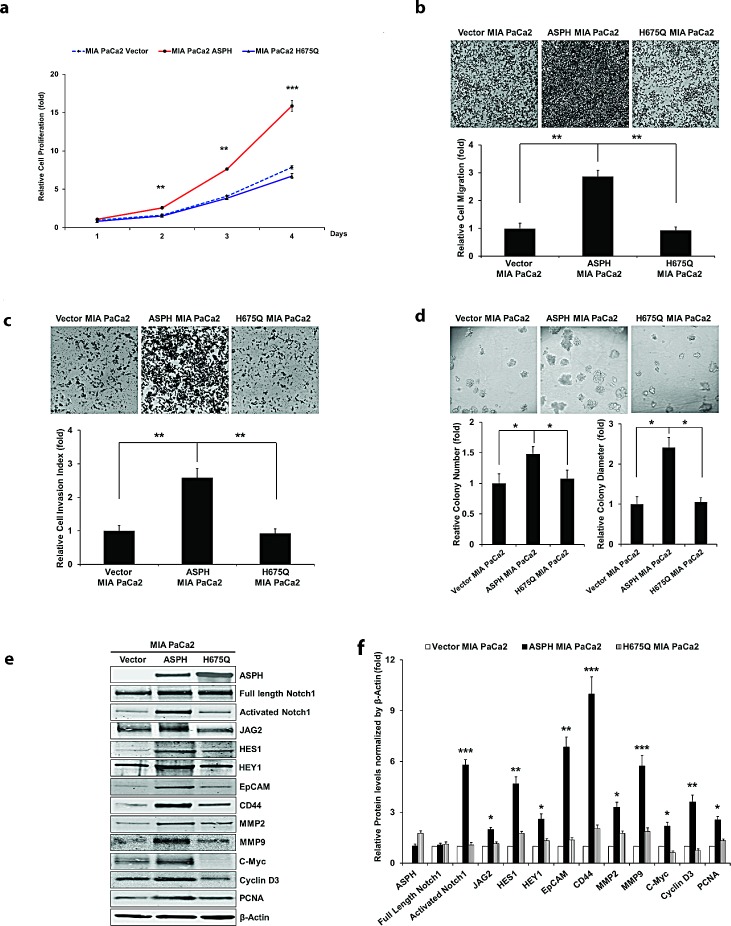
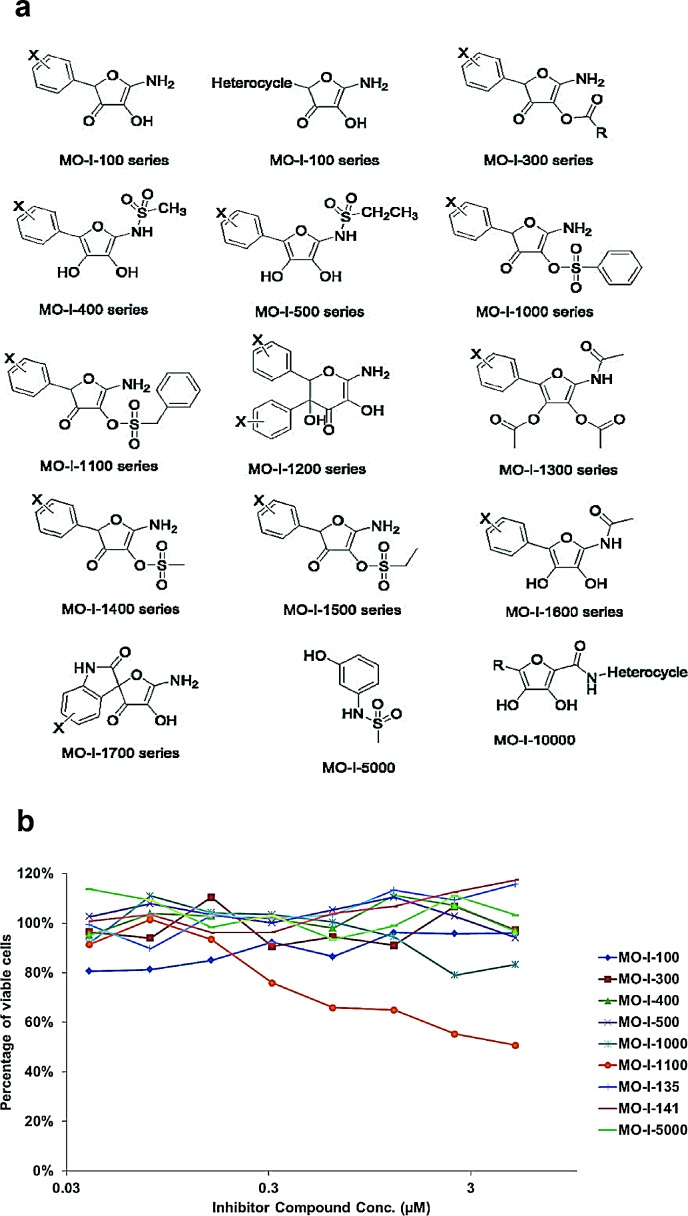
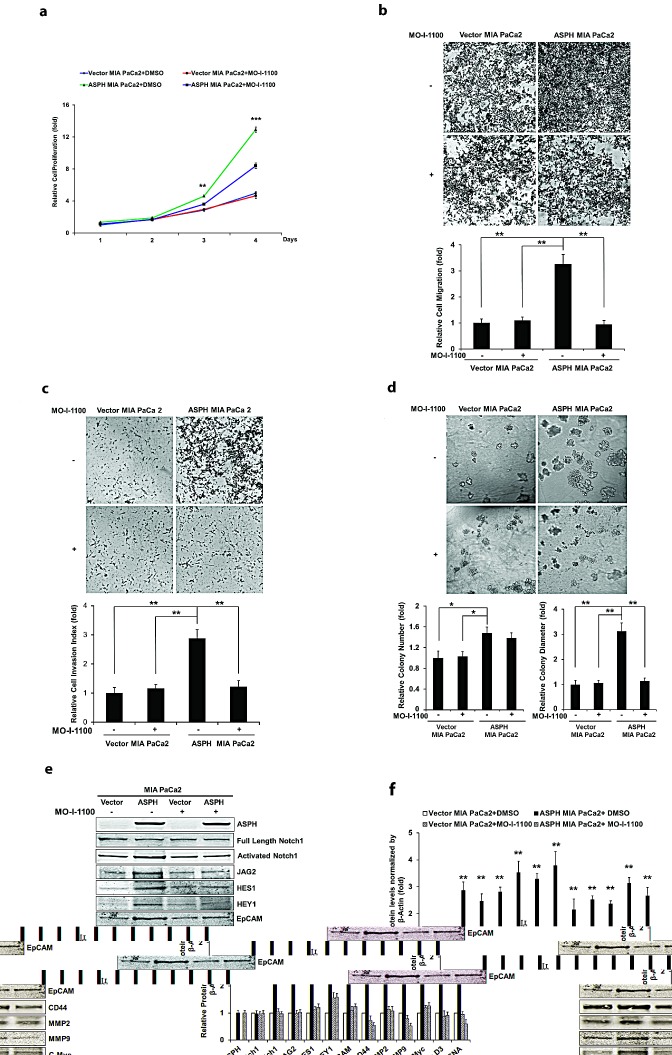
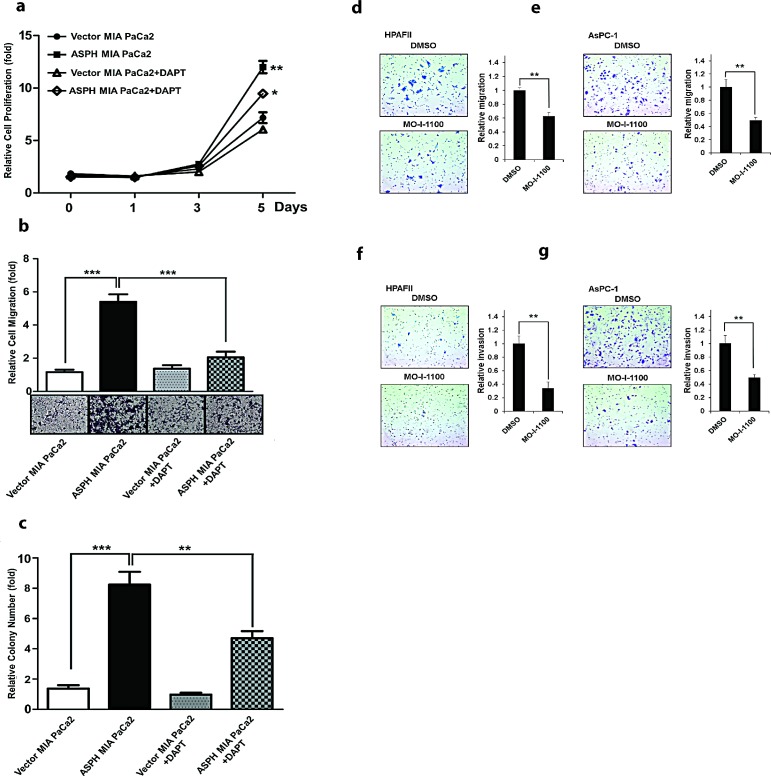
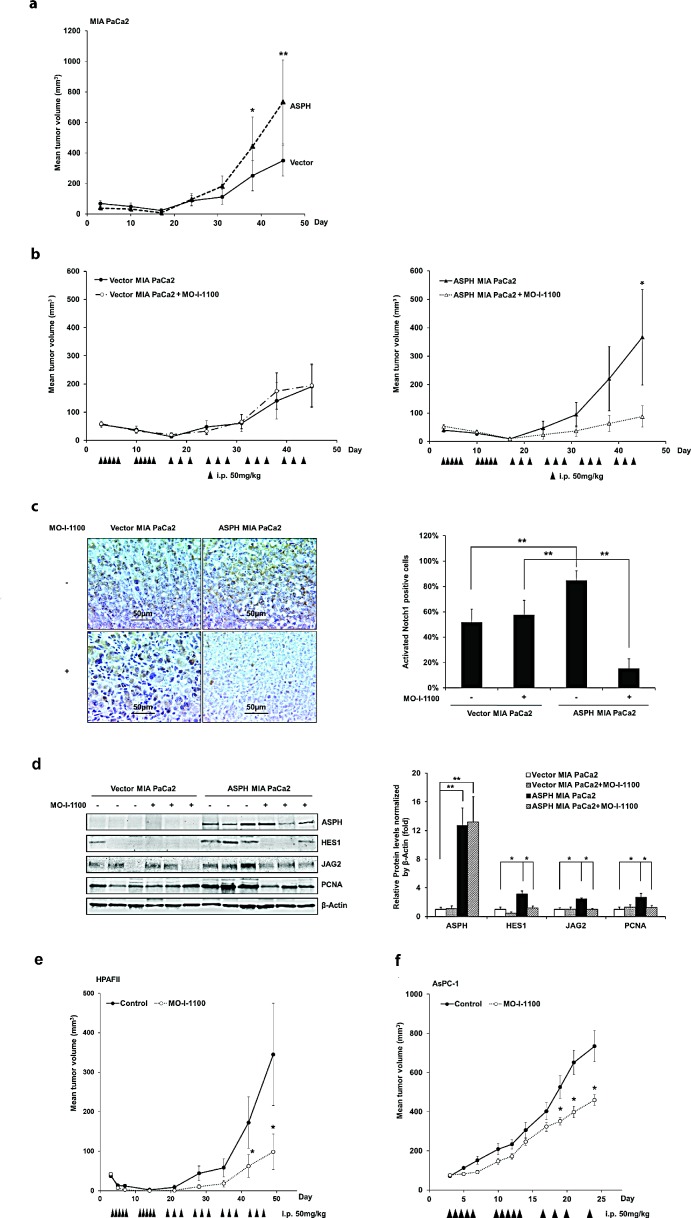
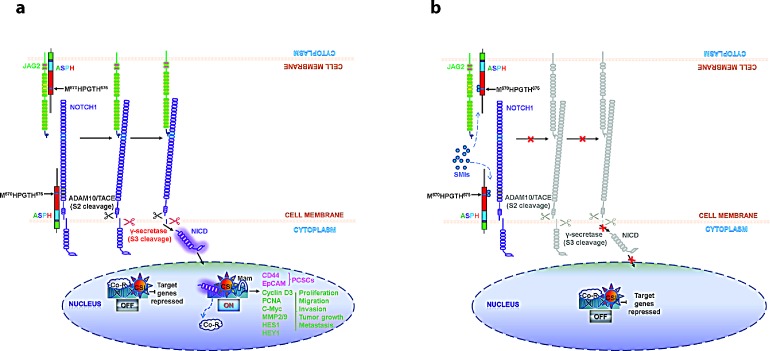
Pancreatic cancer (PC) is one of the leading causes of cancer related deaths due to aggressive progression and metastatic spread. Aspartate β-hydroxylase (ASPH), a cell surface protein that catalyzes the hydroxylation of epidermal growth factor (EGF)-like repeats in Notch receptors and ligands, is highly overexpressed in PC. ASPH upregulation confers a malignant phenotype characterized by enhanced cell proliferation, migration, invasion and colony formation in vitro as well as PC tumor growth in vivo. The transforming properties of ASPH depend on enzymatic activity. ASPH links PC growth factor signaling cascades to Notch activation. A small molecule inhibitor of β-hydroxylase activity was developed and found to reduce PC growth by downregulating the Notch signaling pathway. These findings demonstrate the critical involvement of ASPH in PC growth and progression, provide new insight into the molecular mechanisms leading to tumor development and growth and have important therapeutic implications.
Conflict of interest statement
There is no conflict of interest among the authors.
Figures
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63(1):11–30. - PubMed
-
- Jia S, VanDusen WJ, Diehl RE, Kohl NE, Dixon RA, Elliston KO, Stern AM, Friedman PA. cDNA cloning and expression of bovine aspartyl (asparaginyl) beta-hydroxylase. J Biol Chem. 1992;267(20):14322–14327. - PubMed
-
- Engel J. EGF-like domains in extracellular matrix proteins: localized signals for growth and differentiation? FEBS letters. 1989;251(1-2):1–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
