

Targeting cardiomyocyte Ca2+ homeostasis in heart failure
- PMID: 25483944
- PMCID: PMC4475738
- DOI: 10.2174/138161282104141204124129
Targeting cardiomyocyte Ca2+ homeostasis in heart failure
Abstract
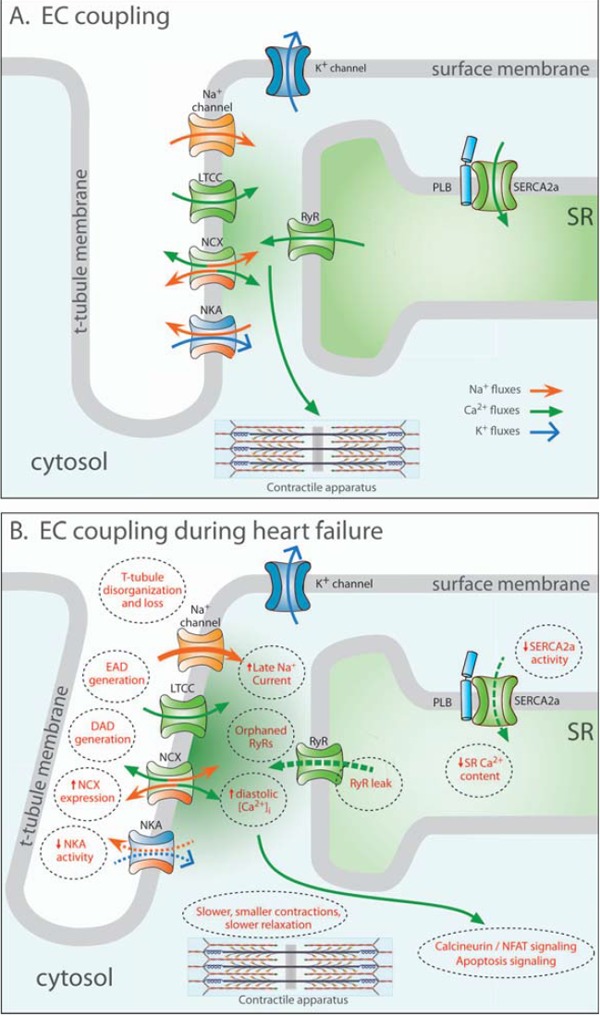
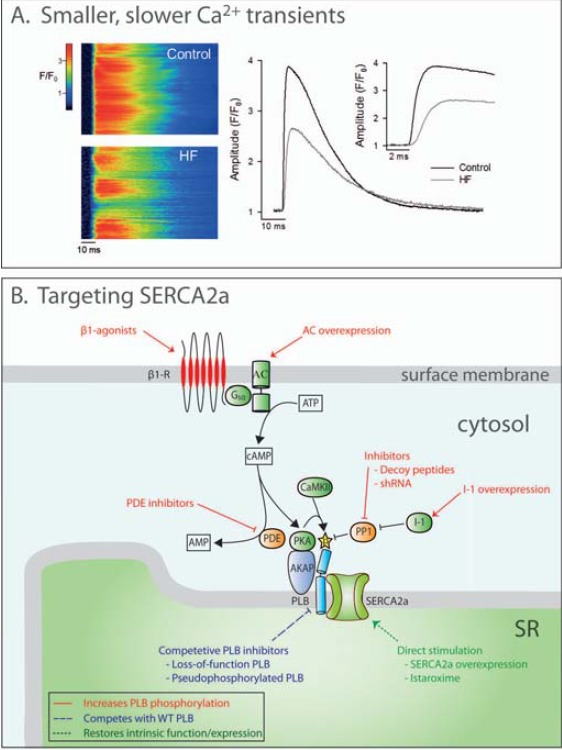
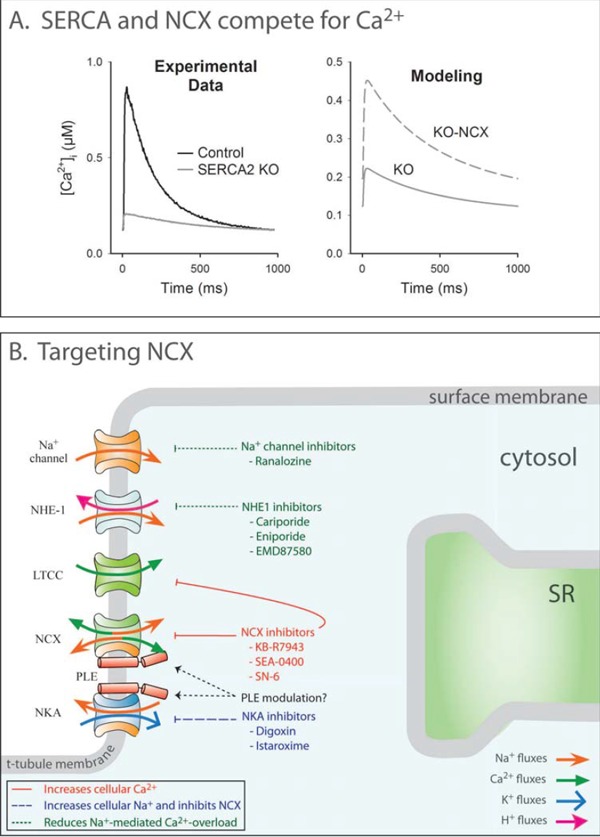
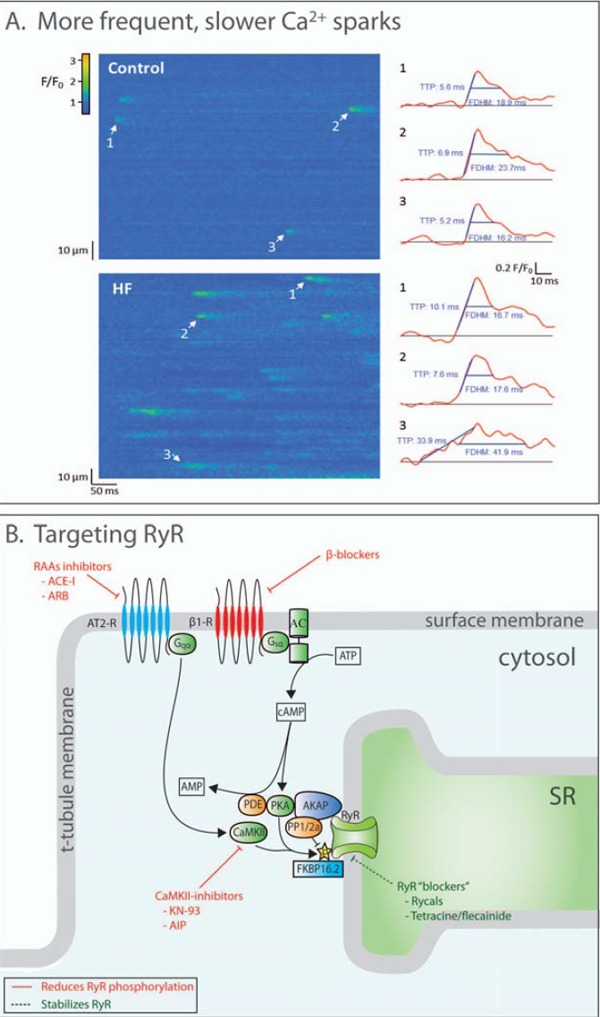
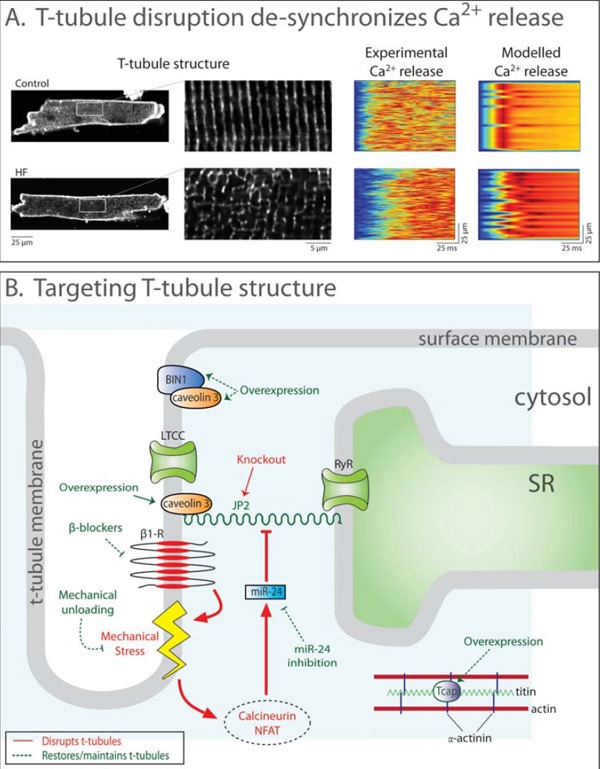
Improved treatments for heart failure patients will require the development of novel therapeutic strategies that target basal disease mechanisms. Disrupted cardiomyocyte Ca(2+) homeostasis is recognized as a major contributor to the heart failure phenotype, as it plays a key role in systolic and diastolic dysfunction, arrhythmogenesis, and hypertrophy and apoptosis signaling. In this review, we outline existing knowledge of the involvement of Ca(2+) homeostasis in these deficits, and identify four promising targets for therapeutic intervention: the sarcoplasmic reticulum Ca(2+) ATPase, the Na(+)-Ca(2+) exchanger, the ryanodine receptor, and t-tubule structure. We discuss experimental data indicating the applicability of these targets that has led to recent and ongoing clinical trials, and suggest future therapeutic approaches.
Figures
References
-
- Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–7. - PubMed
-
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. - PubMed
-
- Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–6. - PubMed
-
- Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D'Hooge J, Sipido K. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–46. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous