

From immunobiology to β-cell biology: the changing perspective on type 1 diabetes
- PMID: 25483958
- PMCID: PMC4594197
- DOI: 10.4161/isl.28778
From immunobiology to β-cell biology: the changing perspective on type 1 diabetes
Abstract
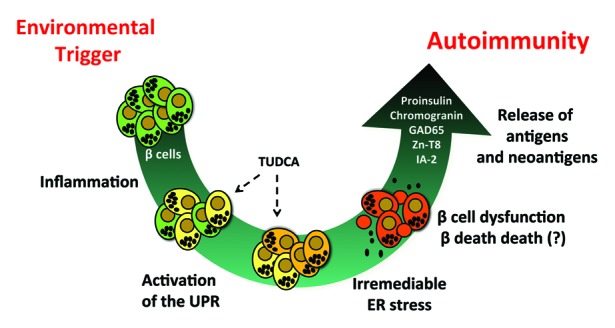
Type 1 Diabetes (T1D) is characterized by the immune mediated destruction of β cells. Clinical studies have focused on drug therapies to modulate autoimmunity, yet none of these interventions has resulted in durable preservation of β-cell function. These findings raise the possibility that initiating or propagating events outside of the immune system should be considered in future efforts to prevent or reverse T1D. An emerging concept suggests that defects inherent to the β cell may trigger autoimmunity. A study by Engin et al. in type 1 diabetic NOD mice suggests that excessive β-cell endoplasmic reticulum stress arising from environmental insults results in abnormal protein synthesis, folding, and/or processing. Administration of the chemical protein folding chaperone TUDCA resulted in recovery of β-cell endoplasmic reticulum function and a diminished incidence of diabetes in NOD mice. We propose here that these data and others support a model whereby an inadequate or defective β-cell endoplasmic reticulum response results in the release of β-cell antigens and neoantigens that initiate autoimmunity. Pharmacologic therapies that either mitigate these early β-cell stressors or enhance the ability of β cells to cope with such stressors may prove to be effective in the prevention or treatment of T1D.
Keywords: ATF6; ER stress; Etiology; Immunology/autoimmunity/immune mechanisms; Islet cell biology/physiology; Pharmacologic agents; autoimmunity; sXBP1; type 1 diabetes; unfolded protein response; β cell.
Figures
Comment on
-
Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes.Sci Transl Med. 2013 Nov 13;5(211):211ra156. doi: 10.1126/scitranslmed.3006534. Sci Transl Med. 2013. PMID: 24225943 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical