

Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway
- PMID: 25484077
- PMCID: PMC4502778
- DOI: 10.4161/15548627.2014.984271
Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway
Abstract
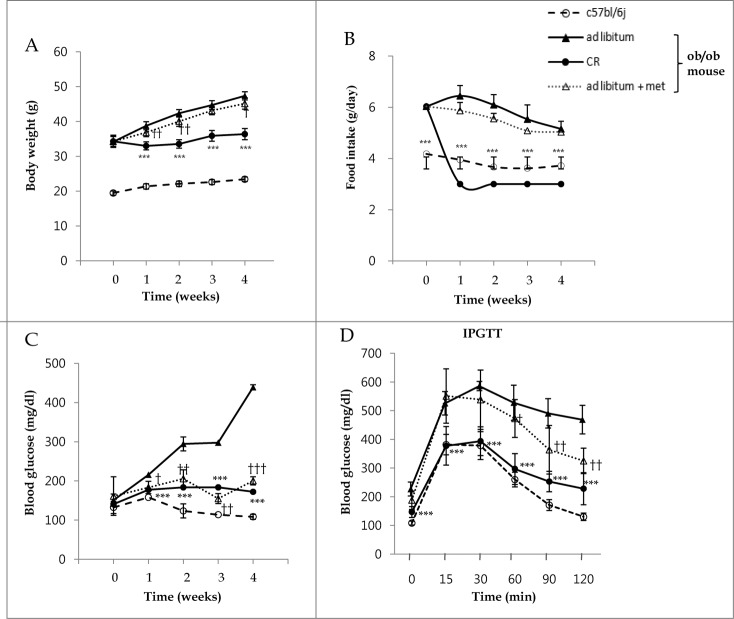
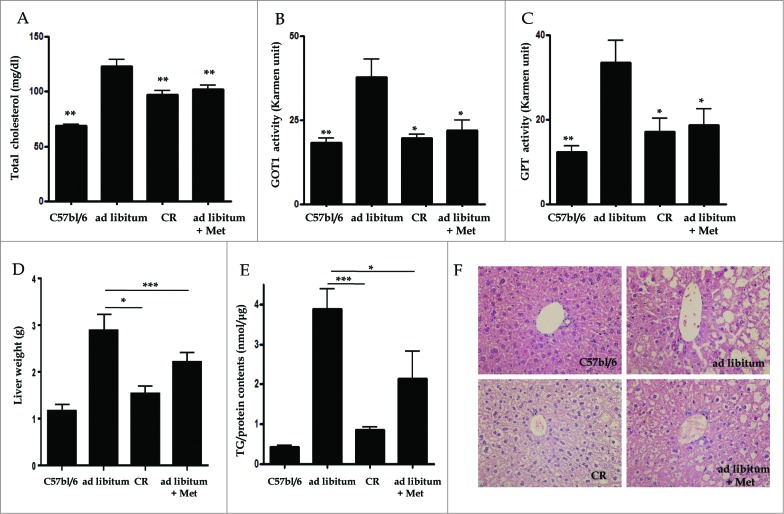
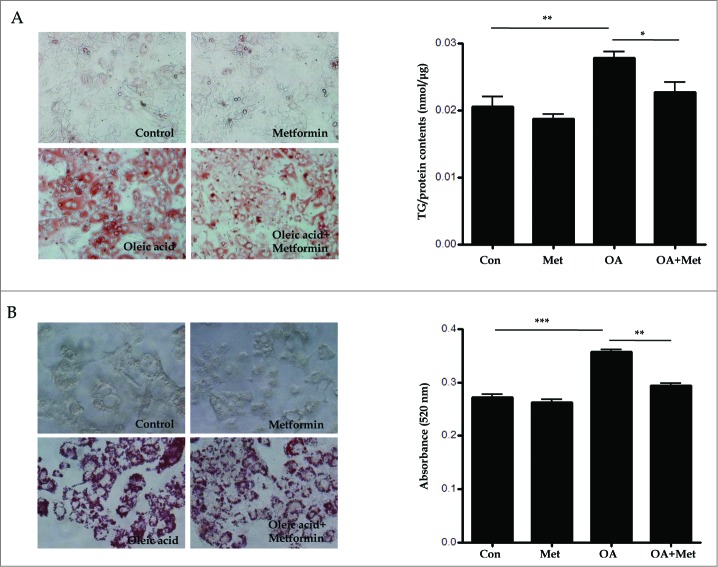
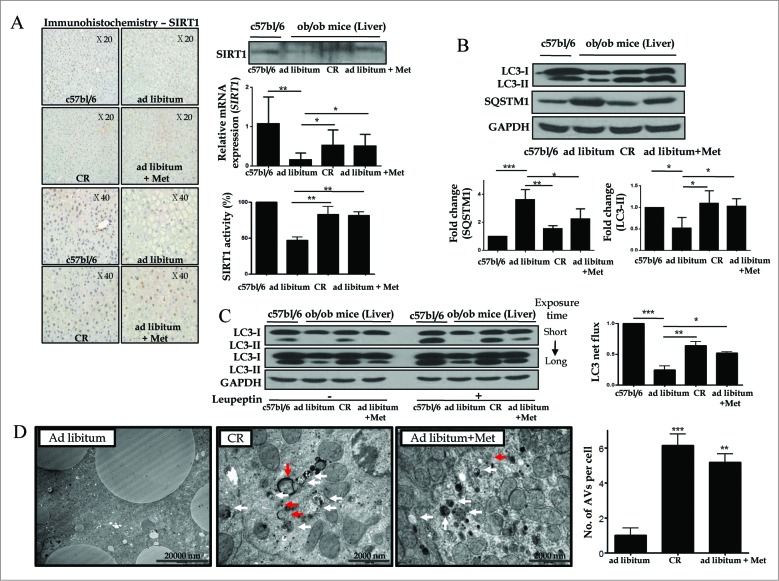
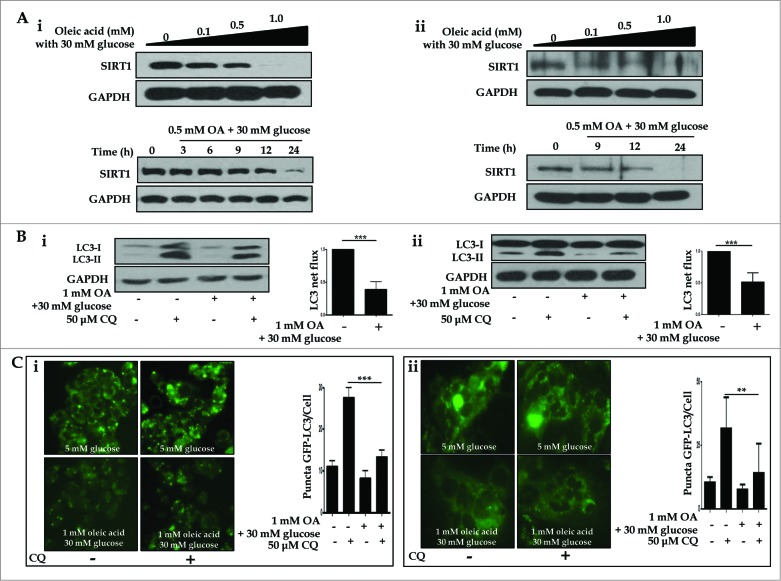
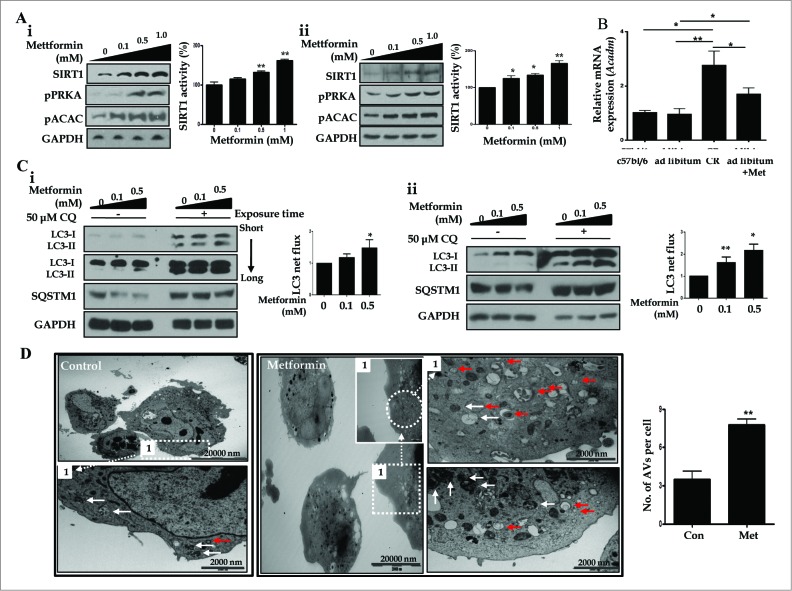
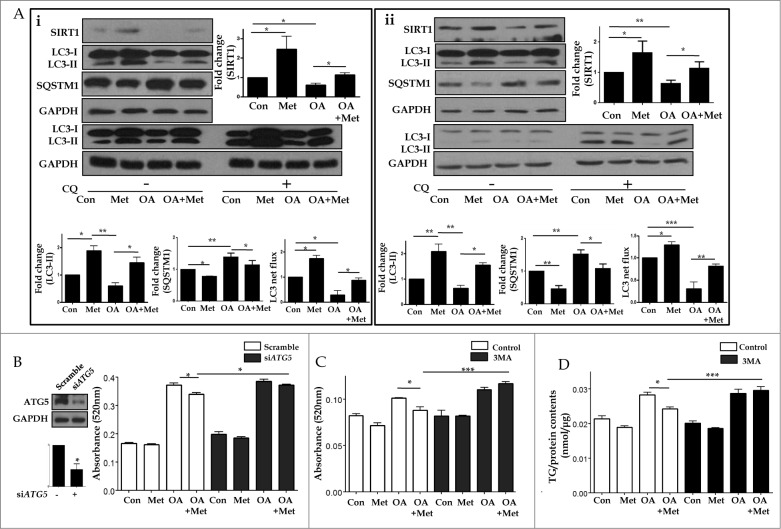
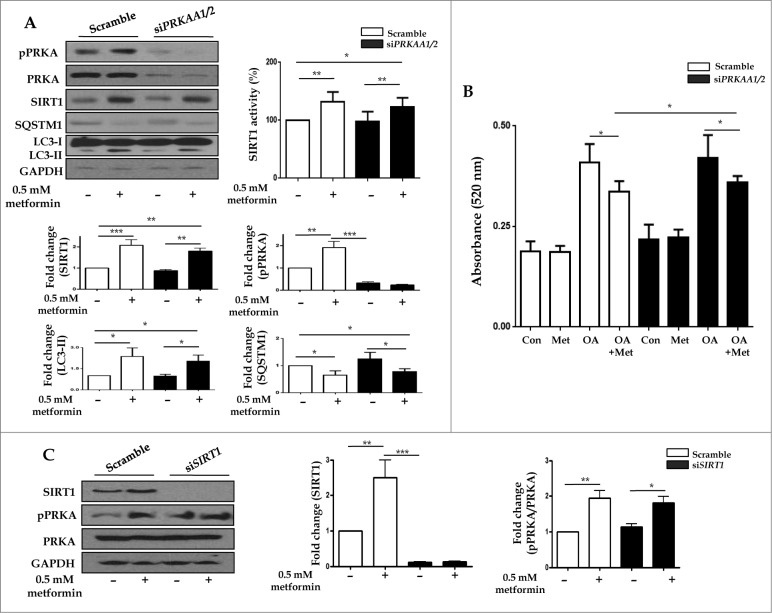
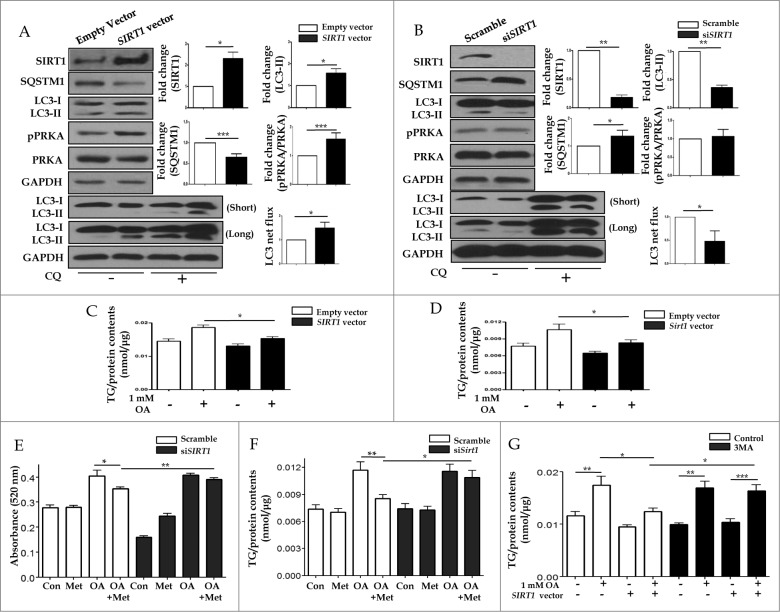
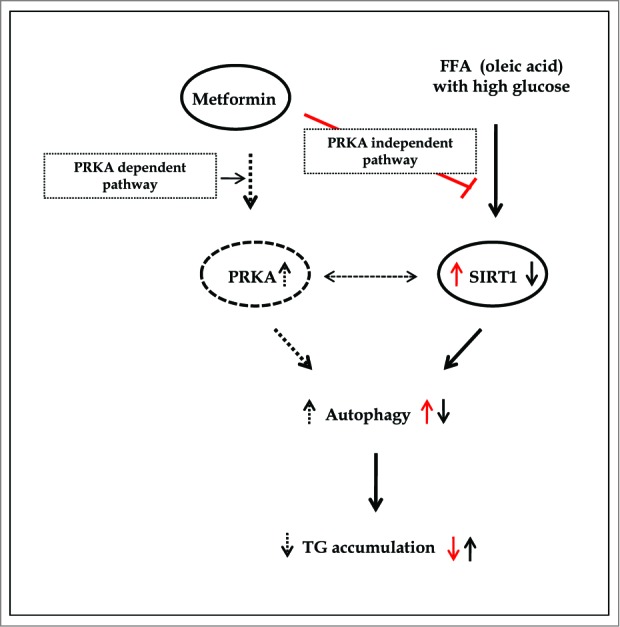
Metformin activates both PRKA and SIRT1. Furthermore, autophagy is induced by either the PRKA-MTOR-ULK1 or SIRT1-FOXO signaling pathways. We aimed to elucidate the mechanism by which metformin alleviates hepatosteatosis by examining the molecular interplay between SIRT1, PRKA, and autophagy. ob/ob mice were divided into 3 groups: one with ad libitum feeding of a standard chow diet, one with 300 mg/kg intraperitoneal metformin injections, and one with 3 g/d caloric restriction (CR) for a period of 4 wk. Primary hepatocytes or HepG2 cells were treated with oleic acid (OA) plus high glucose in the absence or presence of metformin. Both CR and metformin significantly improved body weight and glucose homeostasis, along with hepatic steatosis, in ob/ob mice. Furthermore, CR and metformin both upregulated SIRT1 expression and also stimulated autophagy induction and flux in vivo. Metformin also prevented OA with high glucose-induced suppression of both SIRT1 expression and SIRT1-dependent activation of autophagy machinery, thereby alleviating intracellular lipid accumulation in vitro. Interestingly, metformin treatment upregulated SIRT1 expression and activated PRKA even after siRNA-mediated knockdown of PRKAA1/2 and SIRT1, respectively. Taken together, these results suggest that metformin alleviates hepatic steatosis through PRKA-independent, SIRT1-mediated effects on the autophagy machinery.
Keywords: 3MA, 3-methyladenine; CQ, chloroquine; CR, caloric restriction; GOT1/AST, glutamic-oxaloacetic transaminase 1, soluble; GPT/ALT, glutamic-pyruvate transaminase (alanine aminotransferase); IPGTTs, intraperitoneal glucose tolerance tests; MTOR, mechanistic target of rapamycin; Met, metformin; NAFLD, nonalcoholic fatty liver disease; OA, oleic acid; ORO, Oil Red O; PRKA; PRKA, protein kinase, AMP-activated; SIRT1; SIRT1, sirtuin 1; T-CHO, total cholesterol; TG, triglyceride; autophagy; hepatoseatosis; metformin; siRNA, short interfering RNA.
Figures
References
-
- Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1AMPK pathway via a decrease in hepatic energy state. J Clin Invest 2010; 120:2355-69; PMID:20577053; http://dx.doi.org/10.1172/JCI40671 - DOI - PMC - PubMed
-
- Park HK. Metformin and cancer in type 2 diabetes. Diabetes Metab J 2013; 37:113-6; PMID:23641351; http://dx.doi.org/10.4093/dmj.2013.37.2.113 - DOI - PMC - PubMed
-
- Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 2012; 11:230-41; PMID:22186033; http://dx.doi.org/10.1016/j.arr.2011.12.005 - DOI - PubMed
-
- Pallauf K, Rimbach G. Autophagy, polyphenols and healthy ageing. Ageing Res Rev 2013; 12:237-52; PMID:22504405; http://dx.doi.org/10.1016/j.arr.2012.03.008 - DOI - PubMed
-
- Caton PW, Nayuni NK, Kieswich J, Khan NQ, Yaqoob MM, Corder R. Metformin suppresses hepatic gluconeogenesis through induction of SIRT1 and GCN5. J Endocrinol 2010; 205:97-106; PMID:20093281; http://dx.doi.org/10.1677/JOE-09-0345 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous