

Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells
- PMID: 25485498
- PMCID: PMC4614520
- DOI: 10.4161/15384101.2014.950126
Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells
Abstract
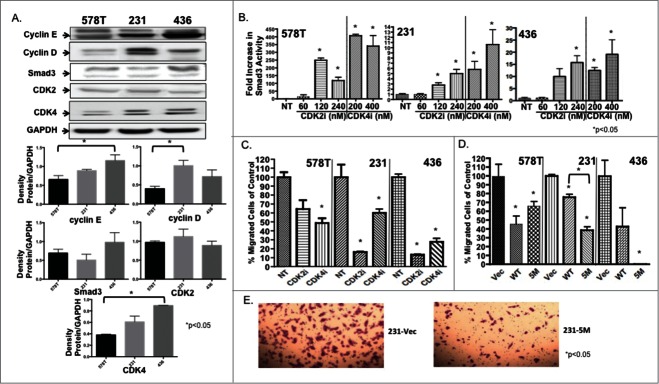
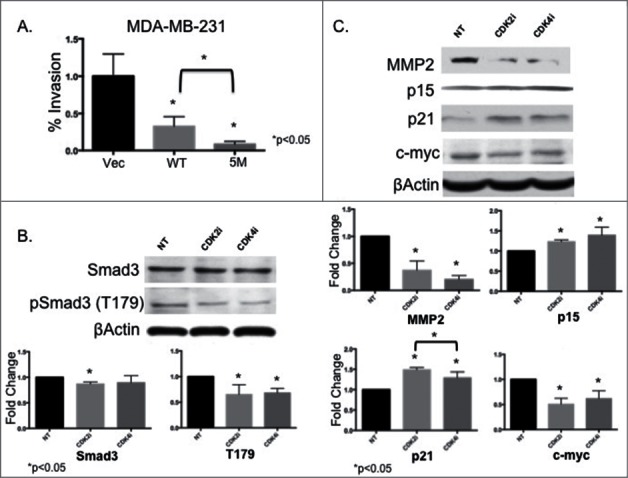
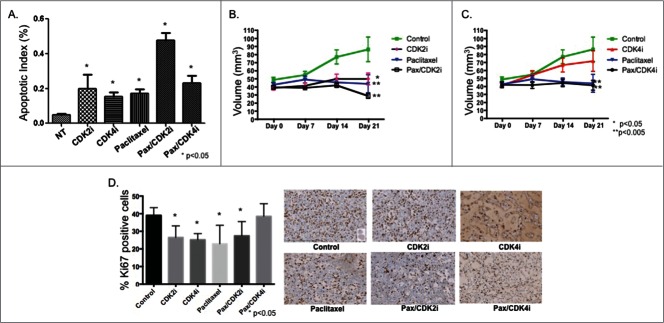
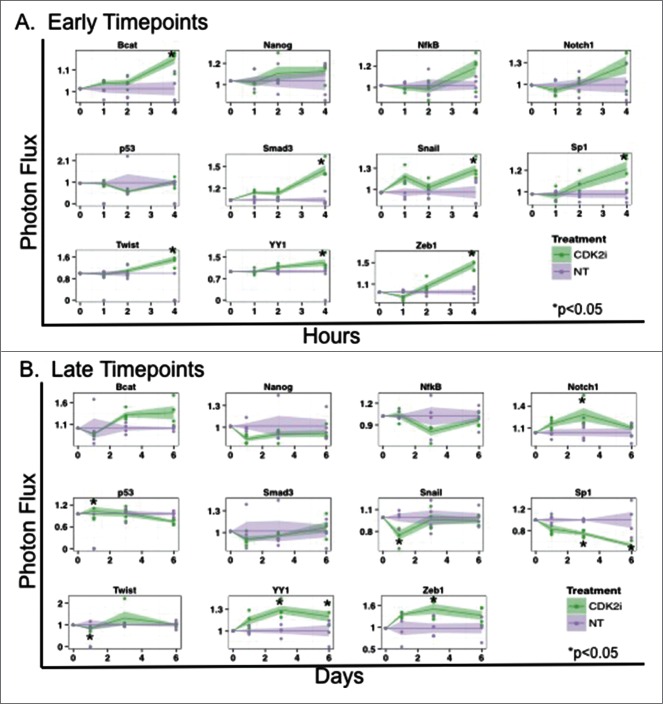
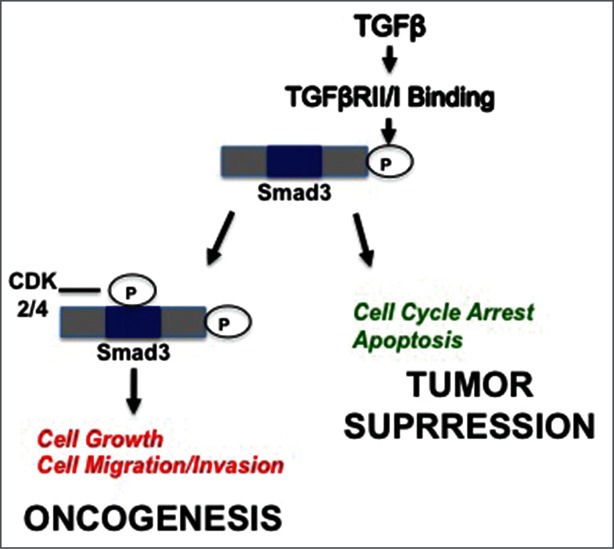
Breast cancer onset and disease progression have been linked to members of the TGFβ superfamily and their downstream signaling components, the Smads. Alterations in Smad3 signaling are associated with the dichotomous role of TGFβ in malignancy, mediating both tumor suppressant and pro-metastatic behaviors. Overexpression of cell cycle regulators, cyclins D and E, renders cyclin-dependent kinases (CDKs) 4/2 hyperactive. Noncanonical phosphorylation of Smad3 by CDK4/2 inhibits tumor suppressant actions of Smad3. We hypothesized that CDK inhibition (CDKi) would restore Smad3 action and help promote cancer cell regression. Treatment of triple-negative breast cancer (TNBC) cell lines (MDA-MB-231, MDA-MB-436, Hs578T) with CDK2i or CDK4i resulted in increased Smad3 activity and decreased cell migration. Transfection with a 5M Smad3 construct containing inhibitory mutations in 5 CDK phosphorylation sites also resulted in decreased TNBC cell migration and invasion. MDA-MB-231 cells treated with CDK2i or CDK4i resulted in decreased Smad3 protein phosphorylation at the CDK phosphorylation T179 site, decreased MMP2 and c-myc expression, and increased p15 and p21 expression. Using a novel transfected cell array, we found that CDK2i treatment decreased activity of the epithelial-to-mesenchymal transition related transcription factors Snail and Twist. In vivo studies in an MDA-MB-231 tumor model showed that individual and combination treatment with paclitaxel and CDK2i resulted in decreased tumor volume and Ki67 staining. Collectively, these data support further investigation of targeted CDK inhibitors as a promising therapeutic strategy for TNBC, a breast cancer subtype with limited treatment options.
Keywords: BCSC, breast cancer stem cells; CDK; CDK, cyclin dependent kinase; CDKi, cyclin dependent kinase inhibitor; CK, cytokeratin; EGFR, epidermal growth factor receptor; EMT, epithelial-mesenchymal transition; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; PR, progesterone receptor; Pin1, peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; Smad3; TNBC, triple negative breast cancer; cyclin; paclitaxel; triple negative breast cancer.
Figures
Similar articles
-
Inhibition of CDK-mediated Smad3 phosphorylation reduces the Pin1-Smad3 interaction and aggressiveness of triple negative breast cancer cells.Cell Cycle. 2017 Aug 3;16(15):1453-1464. doi: 10.1080/15384101.2017.1338988. Epub 2017 Jul 5. Cell Cycle. 2017. PMID: 28678584 Free PMC article.
-
Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells.Mol Cancer Res. 2010 Oct;8(10):1375-87. doi: 10.1158/1541-7786.MCR-09-0537. Epub 2010 Aug 24. Mol Cancer Res. 2010. PMID: 20736297 Free PMC article.
-
Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines.Cell Cycle. 2010 Dec 15;9(24):4900-7. doi: 10.4161/cc.9.24.14158. Epub 2010 Dec 15. Cell Cycle. 2010. PMID: 21150326 Free PMC article.
-
Implications of TGFβ Signaling and CDK Inhibition for the Treatment of Breast Cancer.Cancers (Basel). 2021 Oct 25;13(21):5343. doi: 10.3390/cancers13215343. Cancers (Basel). 2021. PMID: 34771508 Free PMC article. Review.
-
Cyclin-dependent kinase 2 (CDK2) inhibitors and others novel CDK inhibitors (CDKi) in breast cancer: clinical trials, current impact, and future directions.Crit Rev Oncol Hematol. 2024 Apr;196:104324. doi: 10.1016/j.critrevonc.2024.104324. Epub 2024 Mar 8. Crit Rev Oncol Hematol. 2024. PMID: 38462150 Review.
Cited by
-
Cyclin D1, cancer progression, and opportunities in cancer treatment.J Mol Med (Berl). 2016 Dec;94(12):1313-1326. doi: 10.1007/s00109-016-1475-3. Epub 2016 Oct 2. J Mol Med (Berl). 2016. PMID: 27695879 Free PMC article. Review.
-
Structure-Based Design of 2-Aminopurine Derivatives as CDK2 Inhibitors for Triple-Negative Breast Cancer.Front Pharmacol. 2022 May 3;13:864342. doi: 10.3389/fphar.2022.864342. eCollection 2022. Front Pharmacol. 2022. PMID: 35592410 Free PMC article.
-
AT101 Suppresses Gastrointestinal Stromal Tumor Growth and Promotes Apoptosis via YAP/TAZ-CCND1 and FBXW7-MCL1 Axes.Ann Surg Oncol. 2025 Aug;32(8):5991-6004. doi: 10.1245/s10434-025-17247-3. Epub 2025 Mar 27. Ann Surg Oncol. 2025. PMID: 40148719
-
Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors.Pharmaceuticals (Basel). 2024 Mar 1;17(3):326. doi: 10.3390/ph17030326. Pharmaceuticals (Basel). 2024. PMID: 38543112 Free PMC article. Review.
-
A Real-World Evidence Study of CDK4/6 Inhibitor Treatment Patterns and Outcomes in Metastatic Breast Cancer by Germline BRCA Mutation Status.Oncol Ther. 2021 Dec;9(2):575-589. doi: 10.1007/s40487-021-00162-4. Epub 2021 Jul 25. Oncol Ther. 2021. PMID: 34308518 Free PMC article.
References
-
- Guarneri V, Broglio K, Kau SW, Cristofanilli M, Buzdar AU, Valero V, Buchholz T, Meric F, Middleton L, Hortobagyi GN, et al. . Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J Clin Oncol 2006; 24:1037-44; PMID:16505422; http://dx.doi.org/10.1200/JCO.2005.02.6914 - DOI - PubMed
-
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363:1938-48; PMID:21067385; http://dx.doi.org/10.1056/NEJMra1001389 - DOI - PubMed
-
- Fadare O, Tavassoli FA. Clinical and pathologic aspects of basal-like breast cancers. Nat Clin Pract Oncol 2008; 5:149-59; PMID:18212769; http://dx.doi.org/10.1038/ncponc1038 - DOI - PubMed
-
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. . Molecular portraits of human breast tumours. Nature 2000; 406:747-52; PMID:10963602; http://dx.doi.org/10.1038/35021093 - DOI - PubMed
-
- Velasco-Velazquez MA, Li Z, Casimiro M, Loro E, Homsi N, Pestell RG. Examining the role of cyclin D1 in breast cancer. Future Oncol 2011; 7:753-65; PMID:21675838; http://dx.doi.org/10.2217/fon.11.56 - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous