

Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes
- PMID: 25488666
- PMCID: PMC4317045
- DOI: 10.1074/jbc.M114.628628
Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes
Abstract
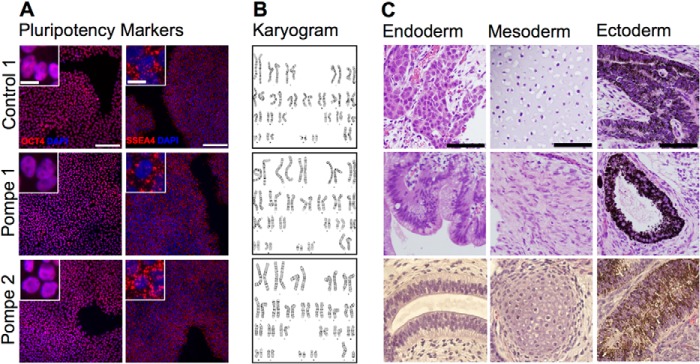
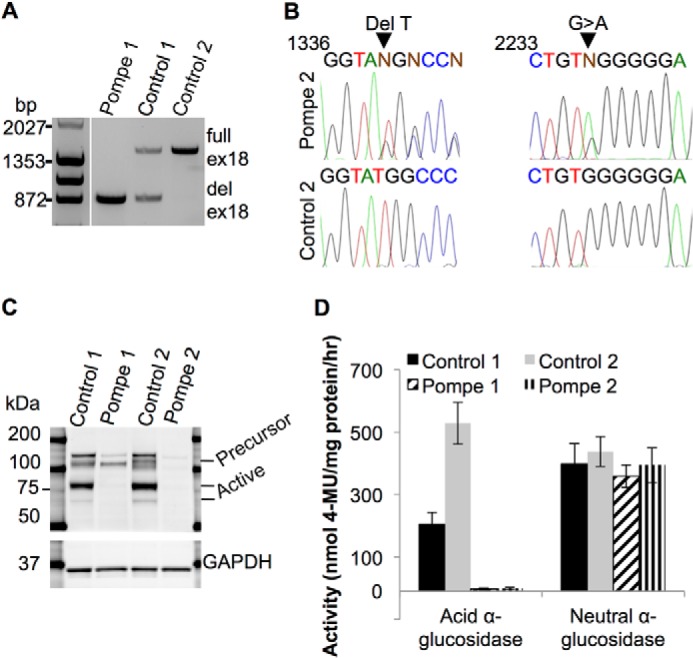
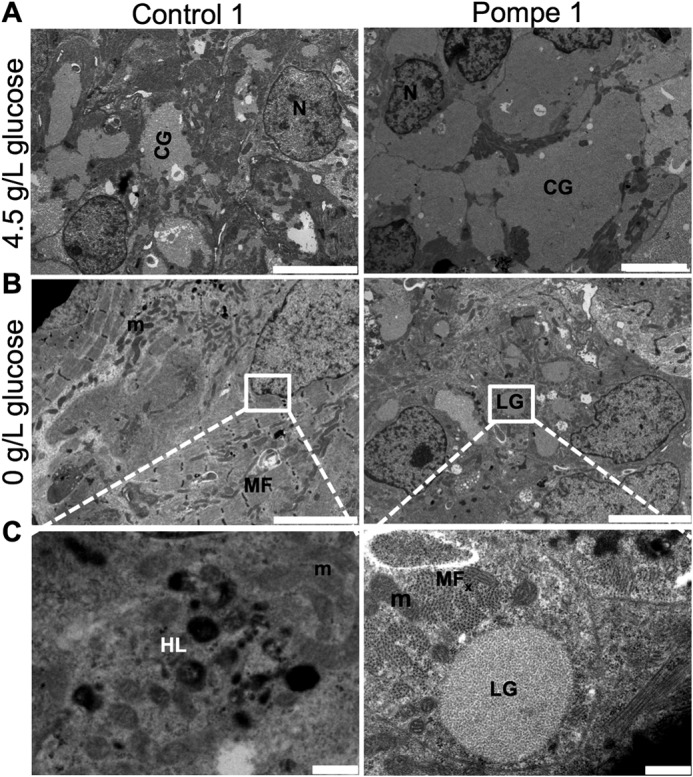
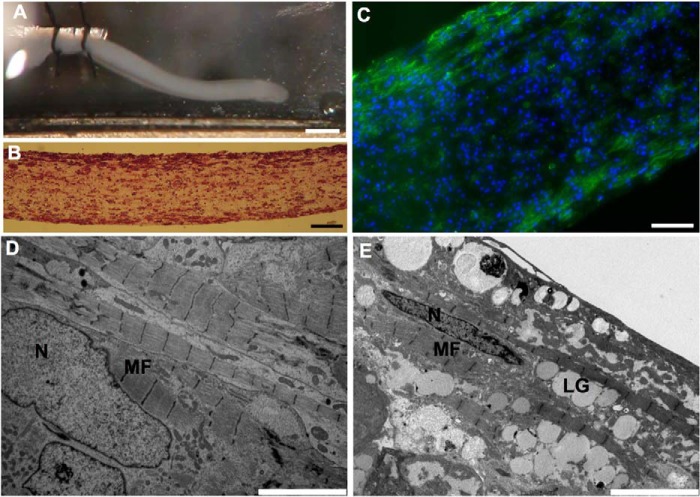
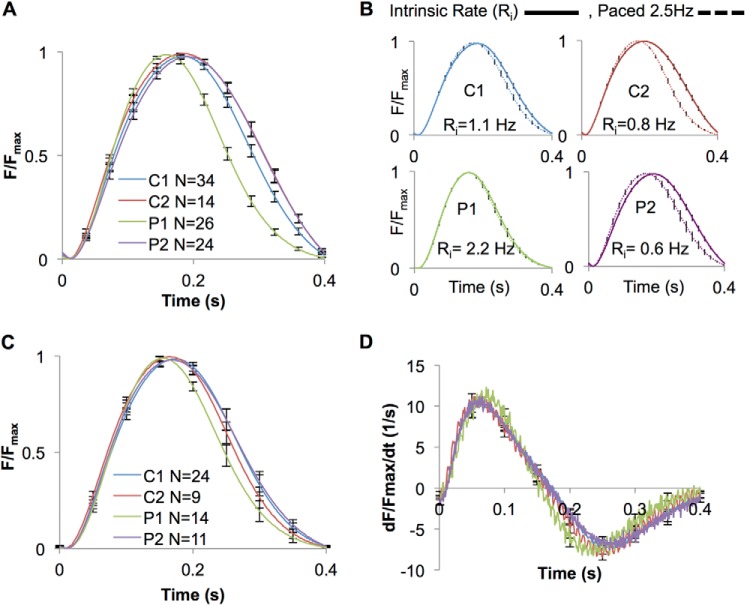
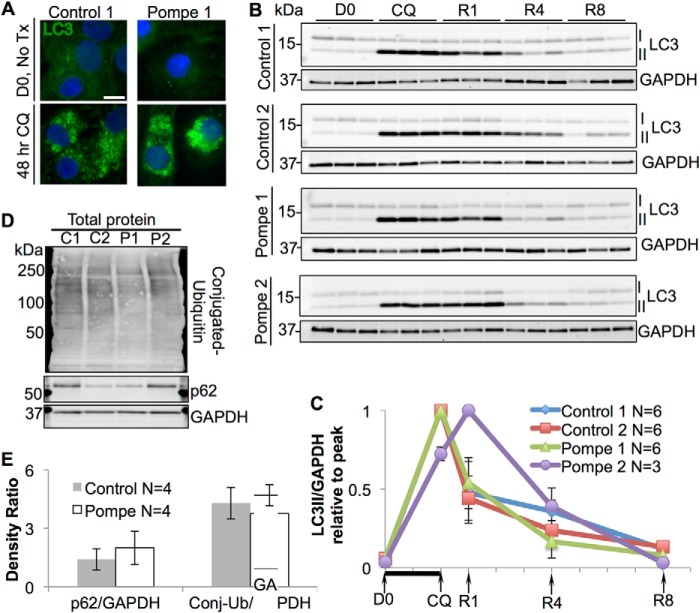
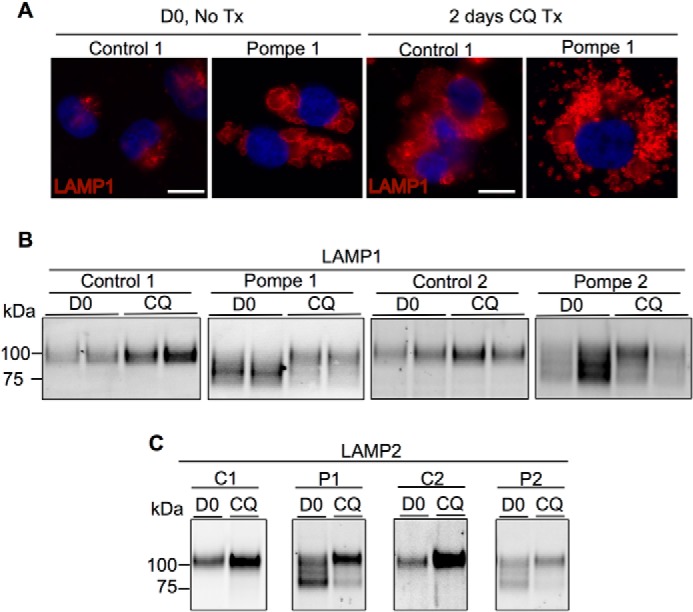
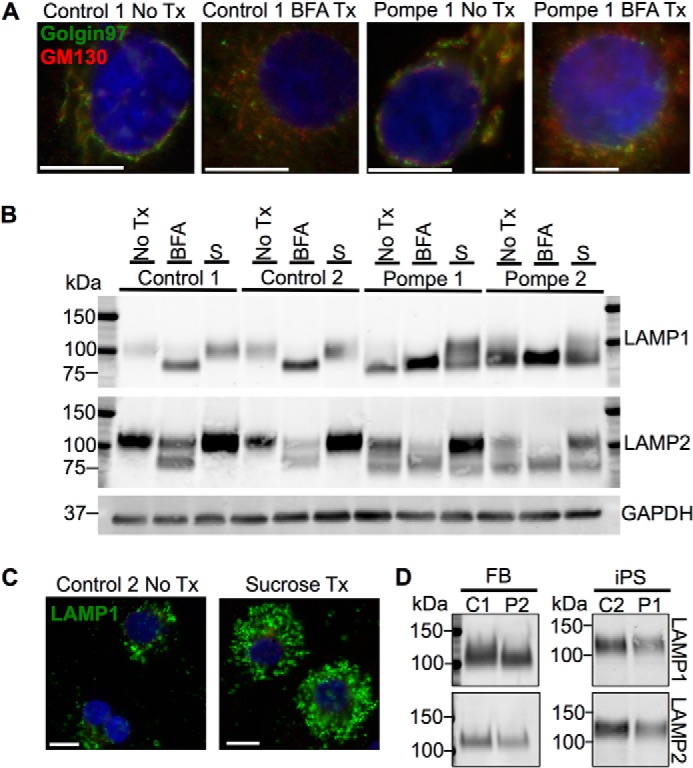
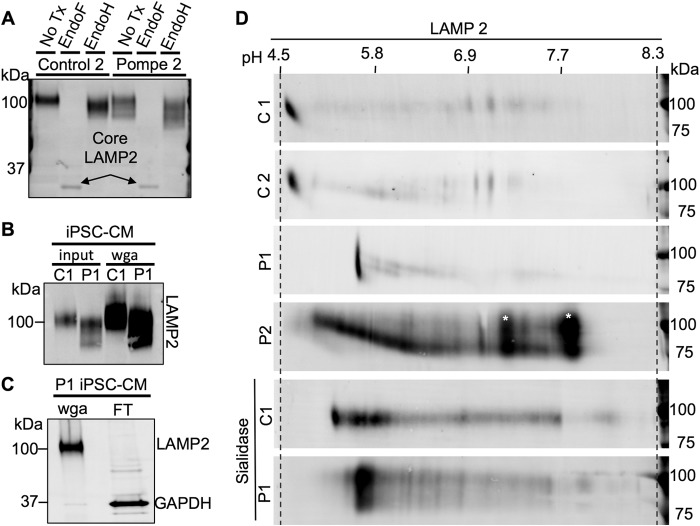
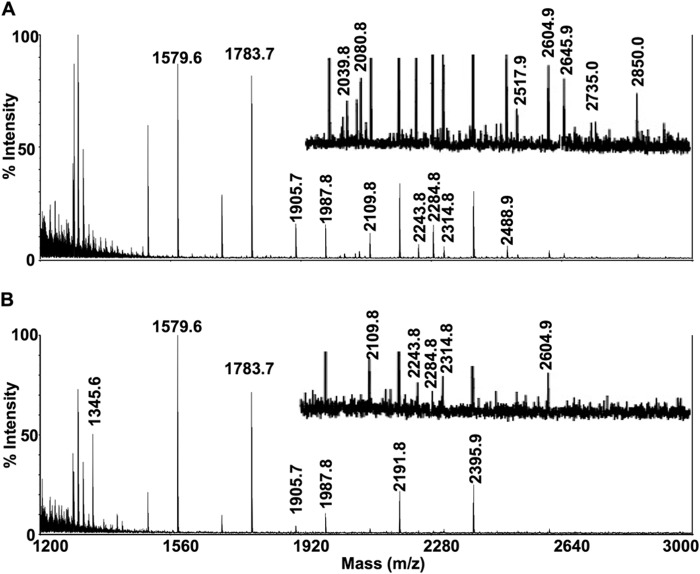
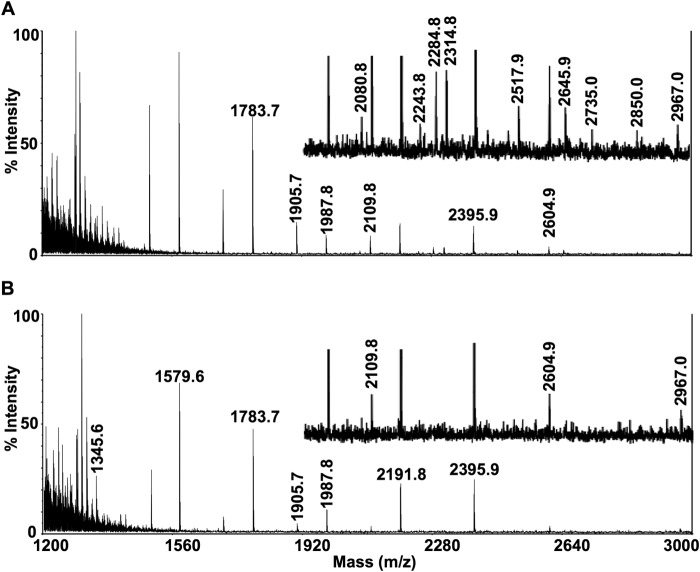
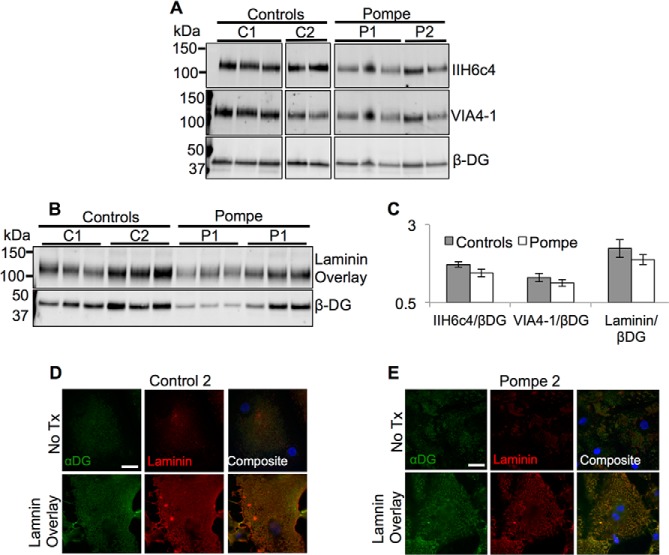
Infantile-onset Pompe disease is an autosomal recessive disorder caused by the complete loss of lysosomal glycogen-hydrolyzing enzyme acid α-glucosidase (GAA) activity, which results in lysosomal glycogen accumulation and prominent cardiac and skeletal muscle pathology. The mechanism by which loss of GAA activity causes cardiomyopathy is poorly understood. We reprogrammed fibroblasts from patients with infantile-onset Pompe disease to generate induced pluripotent stem (iPS) cells that were differentiated to cardiomyocytes (iPSC-CM). Pompe iPSC-CMs had undetectable GAA activity and pathognomonic glycogen-filled lysosomes. Nonetheless, Pompe and control iPSC-CMs exhibited comparable contractile properties in engineered cardiac tissue. Impaired autophagy has been implicated in Pompe skeletal muscle; however, control and Pompe iPSC-CMs had comparable clearance rates of LC3-II-detected autophagosomes. Unexpectedly, the lysosome-associated membrane proteins, LAMP1 and LAMP2, from Pompe iPSC-CMs demonstrated higher electrophoretic mobility compared with control iPSC-CMs. Brefeldin A induced disruption of the Golgi in control iPSC-CMs reproduced the higher mobility forms of the LAMPs, suggesting that Pompe iPSC-CMs produce LAMPs lacking appropriate glycosylation. Isoelectric focusing studies revealed that LAMP2 has a more alkaline pI in Pompe compared with control iPSC-CMs due largely to hyposialylation. MALDI-TOF-MS analysis of N-linked glycans demonstrated reduced diversity of multiantennary structures and the major presence of a trimannose complex glycan precursor in Pompe iPSC-CMs. These data suggest that Pompe cardiomyopathy has a glycan processing abnormality and thus shares features with hypertrophic cardiomyopathies observed in the congenital disorders of glycosylation.
Keywords: Autophagy; Cardiomyopathy; Golgi; Induced Pluripotent Stem Cell (iPSC); Lysosomal Glycoprotein; N-linked Glycosylation; Pompe Disease; Tissue Engineering.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Metabolomic Profiling of Pompe Disease-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals That Oxidative Stress Is Associated with Cardiac and Skeletal Muscle Pathology.Stem Cells Transl Med. 2017 Jan;6(1):31-39. doi: 10.5966/sctm.2015-0409. Epub 2016 Aug 18. Stem Cells Transl Med. 2017. PMID: 28170191 Free PMC article.
-
A Skeletal Muscle Model of Infantile-onset Pompe Disease with Patient-specific iPS Cells.Sci Rep. 2017 Oct 18;7(1):13473. doi: 10.1038/s41598-017-14063-y. Sci Rep. 2017. PMID: 29044175 Free PMC article.
-
Using human Pompe disease-induced pluripotent stem cell-derived neural cells to identify compounds with therapeutic potential.Hum Mol Genet. 2019 Dec 1;28(23):3880-3894. doi: 10.1093/hmg/ddz218. Hum Mol Genet. 2019. PMID: 31518394
-
Failure of Autophagy in Pompe Disease.Biomolecules. 2024 May 13;14(5):573. doi: 10.3390/biom14050573. Biomolecules. 2024. PMID: 38785980 Free PMC article. Review.
-
The genotype-phenotype correlation in Pompe disease.Am J Med Genet C Semin Med Genet. 2012 Feb 15;160C(1):59-68. doi: 10.1002/ajmg.c.31318. Epub 2012 Jan 17. Am J Med Genet C Semin Med Genet. 2012. PMID: 22253258 Review.
Cited by
-
Human Induced Pluripotent Stem Cells as a Platform for Personalized and Precision Cardiovascular Medicine.Physiol Rev. 2016 Jul;96(3):1093-126. doi: 10.1152/physrev.00036.2015. Physiol Rev. 2016. PMID: 27335446 Free PMC article. Review.
-
Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes-a systematic review.Pflugers Arch. 2019 May;471(5):755-768. doi: 10.1007/s00424-018-2214-0. Epub 2018 Oct 15. Pflugers Arch. 2019. PMID: 30324321 Free PMC article.
-
From Acid Alpha-Glucosidase Deficiency to Autophagy: Understanding the Bases of POMPE Disease.Int J Mol Sci. 2023 Aug 5;24(15):12481. doi: 10.3390/ijms241512481. Int J Mol Sci. 2023. PMID: 37569856 Free PMC article.
-
hiPSC-Derived Cardiac Tissue for Disease Modeling and Drug Discovery.Int J Mol Sci. 2020 Nov 24;21(23):8893. doi: 10.3390/ijms21238893. Int J Mol Sci. 2020. PMID: 33255277 Free PMC article. Review.
-
Three-dimensional tissue-engineered human skeletal muscle model of Pompe disease.Commun Biol. 2021 May 5;4(1):524. doi: 10.1038/s42003-021-02059-4. Commun Biol. 2021. PMID: 33953320 Free PMC article.
References
-
- van der Ploeg A. T., Reuser A. J. (2008) Pompe's disease. Lancet 372, 1342–1353 - PubMed
-
- Kishnani P. S., Hwu W. L., Mandel H., Nicolino M., Yong F., Corzo D., and Infantile-Onset Pompe Disease Natural History Study Group (2006) A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 148, 671–676 - PubMed
-
- Kishnani P. S., Goldenberg P. C., DeArmey S. L., Heller J., Benjamin D., Young S., Bali D., Smith S. A., Li J. S., Mandel H., Koeberl D., Rosenberg A., Chen Y. T. (2010) Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 99, 26–33 - PMC - PubMed
-
- Chen L. R., Chen C. A., Chiu S. N., Chien Y. H., Lee N. C., Lin M. T., Hwu W. L., Wang J. K., Wu M. H. (2009) Reversal of cardiac dysfunction after enzyme replacement in patients with infantile-onset Pompe disease. J. Pediatr. 155, 271–275 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
