

Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K
- PMID: 25488983
- PMCID: PMC4267241
- DOI: 10.1084/jem.20141759
Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K
Abstract
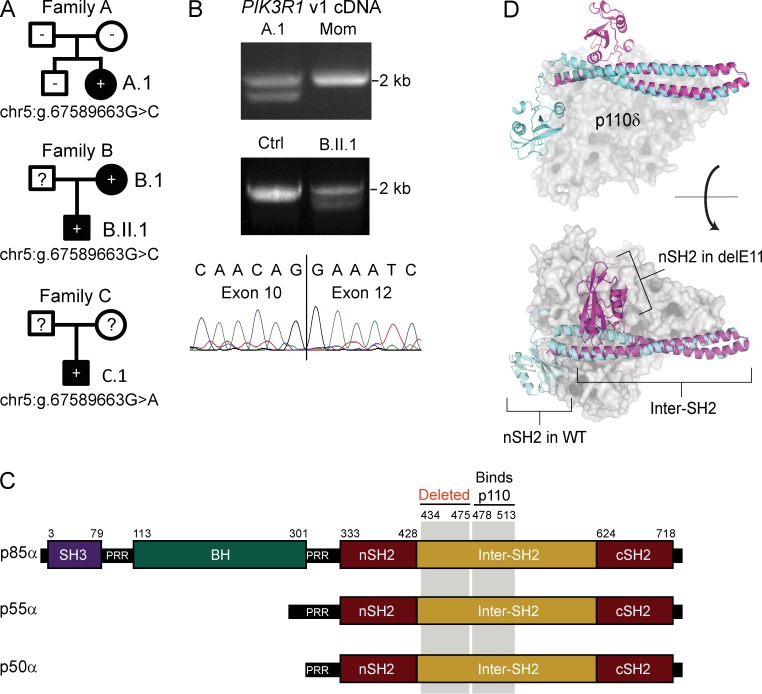
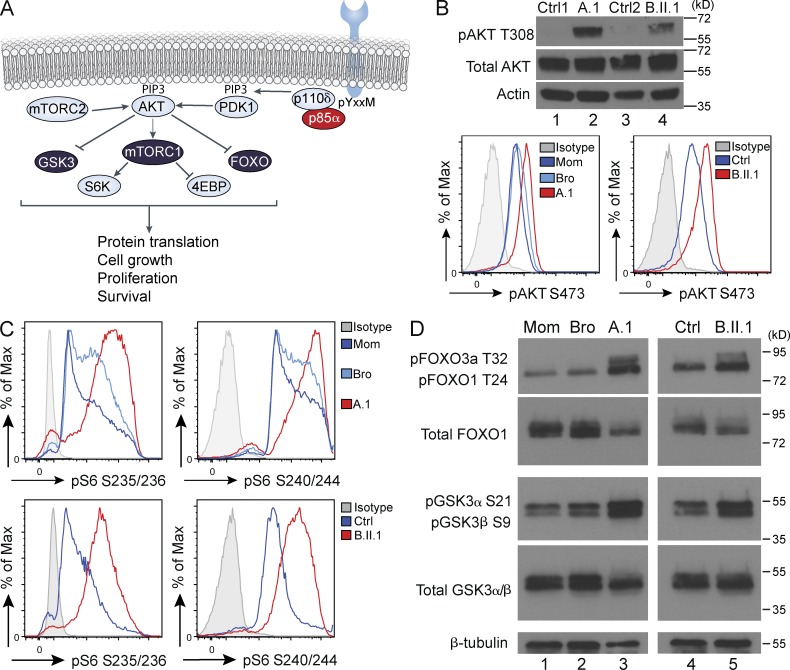
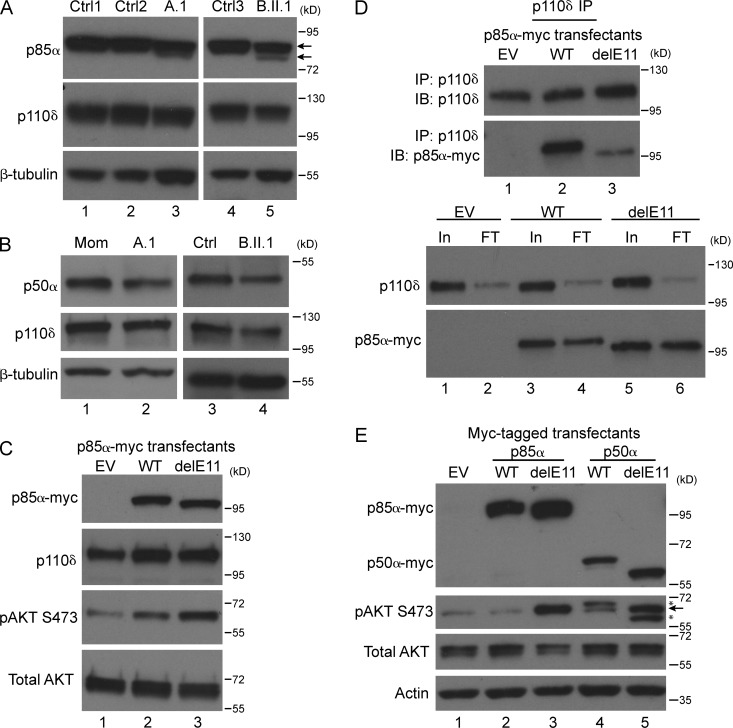
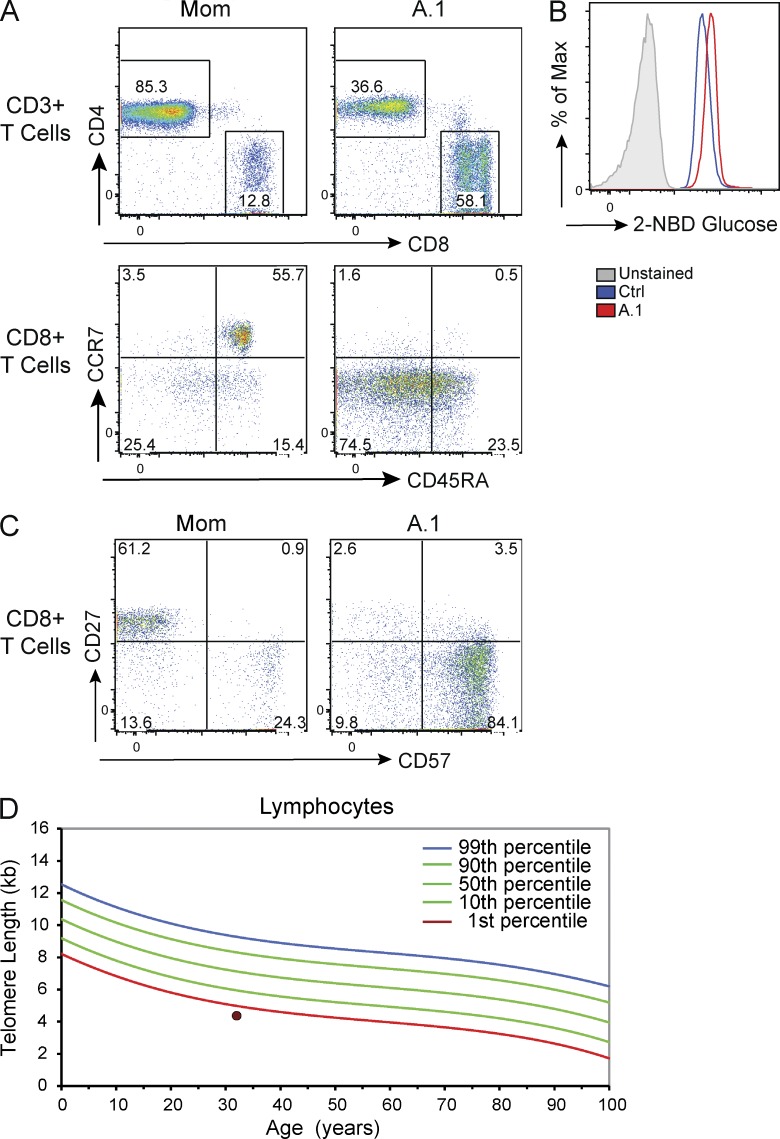
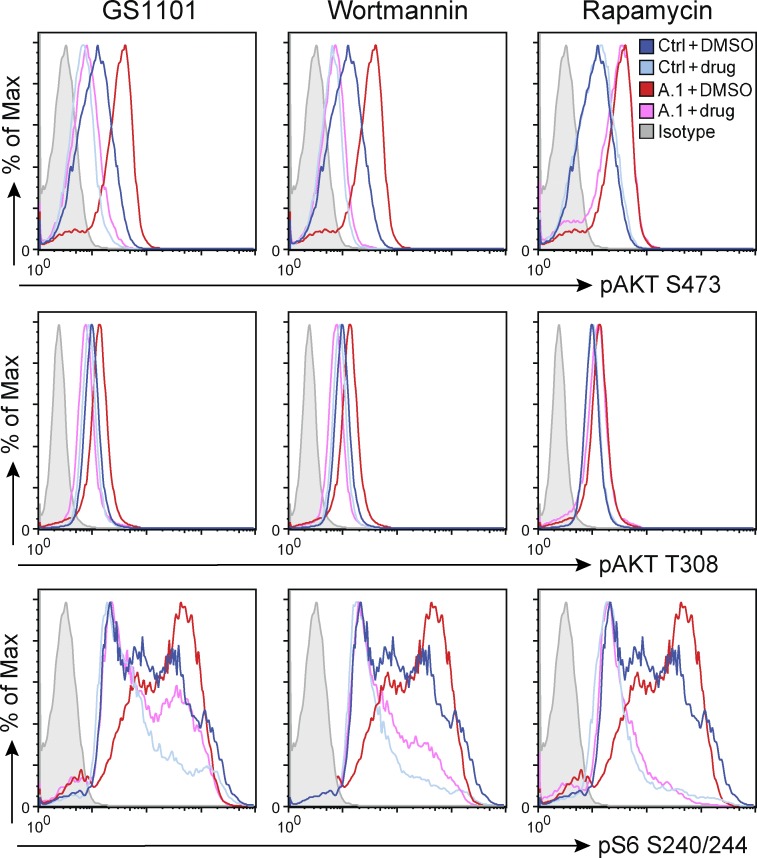
Class IA phosphatidylinositol 3-kinases (PI3K), which generate PIP3 as a signal for cell growth and proliferation, exist as an intracellular complex of a catalytic subunit bound to a regulatory subunit. We and others have previously reported that heterozygous mutations in PIK3CD encoding the p110δ catalytic PI3K subunit cause a unique disorder termed p110δ-activating mutations causing senescent T cells, lymphadenopathy, and immunodeficiency (PASLI) disease. We report four patients from three families with a similar disease who harbor a recently reported heterozygous splice site mutation in PIK3R1, which encodes the p85α, p55α, and p50α regulatory PI3K subunits. These patients suffer from recurrent sinopulmonary infections and lymphoproliferation, exhibit hyperactive PI3K signaling, and have prominent expansion and skewing of peripheral blood CD8(+) T cells toward terminally differentiated senescent effector cells with short telomeres. The PIK3R1 splice site mutation causes skipping of an exon, corresponding to loss of amino acid residues 434-475 in the inter-SH2 domain. The mutant p85α protein is expressed at low levels in patient cells and activates PI3K signaling when overexpressed in T cells from healthy subjects due to qualitative and quantitative binding changes in the p85α-p110δ complex and failure of the C-terminal region to properly inhibit p110δ catalytic activity.
Figures
References
-
- Bárcena C., Quesada V., De Sandre-Giovannoli A., Puente D.A., Fernández-Toral J., Sigaudy S., Baban A., Lévy N., Velasco G., and López-Otín C.. 2014. Exome sequencing identifies a novel mutation in PIK3R1 as the cause of SHORT syndrome. BMC Med. Genet. 15:51 10.1186/1471-2350-15-51 - DOI - PMC - PubMed
-
- Brown J.R., Byrd J.C., Coutre S.E., Benson D.M., Flinn I.W., Wagner-Johnston N.D., Spurgeon S.E., Kahl B.S., Bello C., Webb H.K., et al. 2014. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 123:3390–3397 10.1182/blood-2013-11-535047 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
