

Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling
- PMID: 25489073
- PMCID: PMC4280597
- DOI: 10.1073/pnas.1414665111
Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling
Abstract
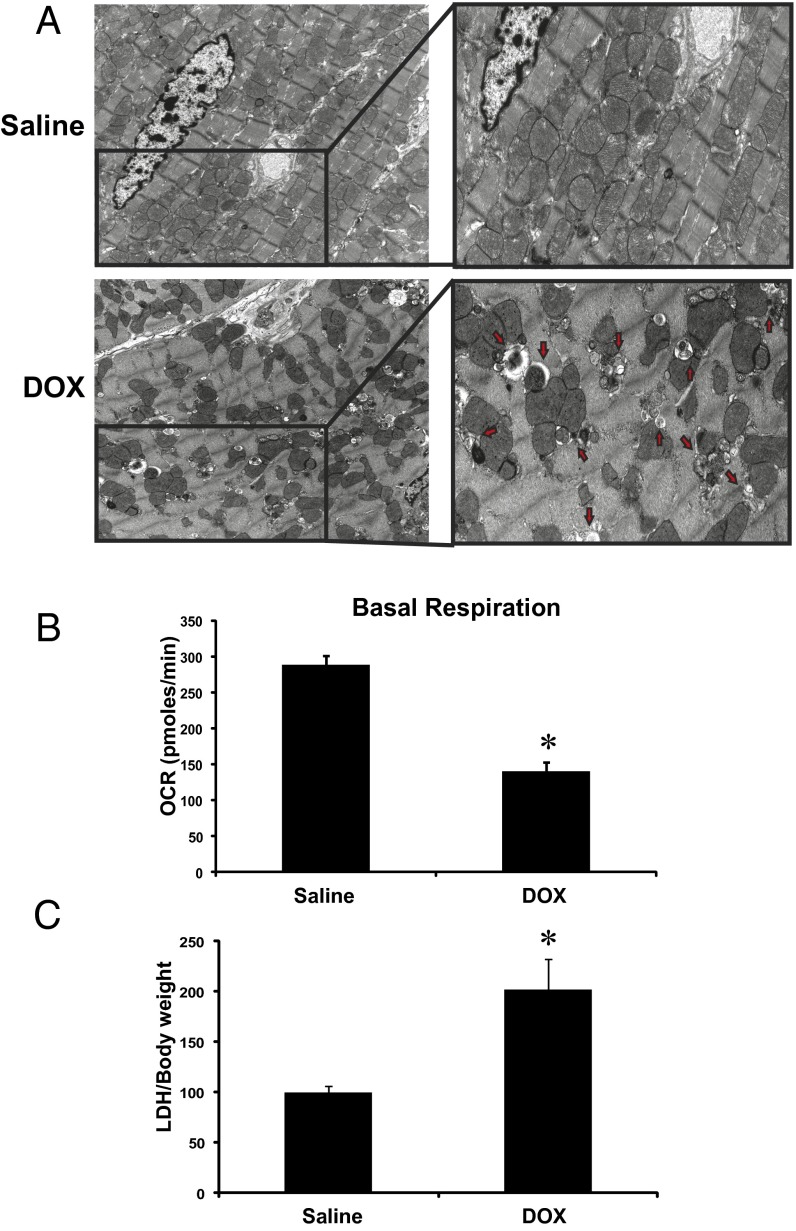
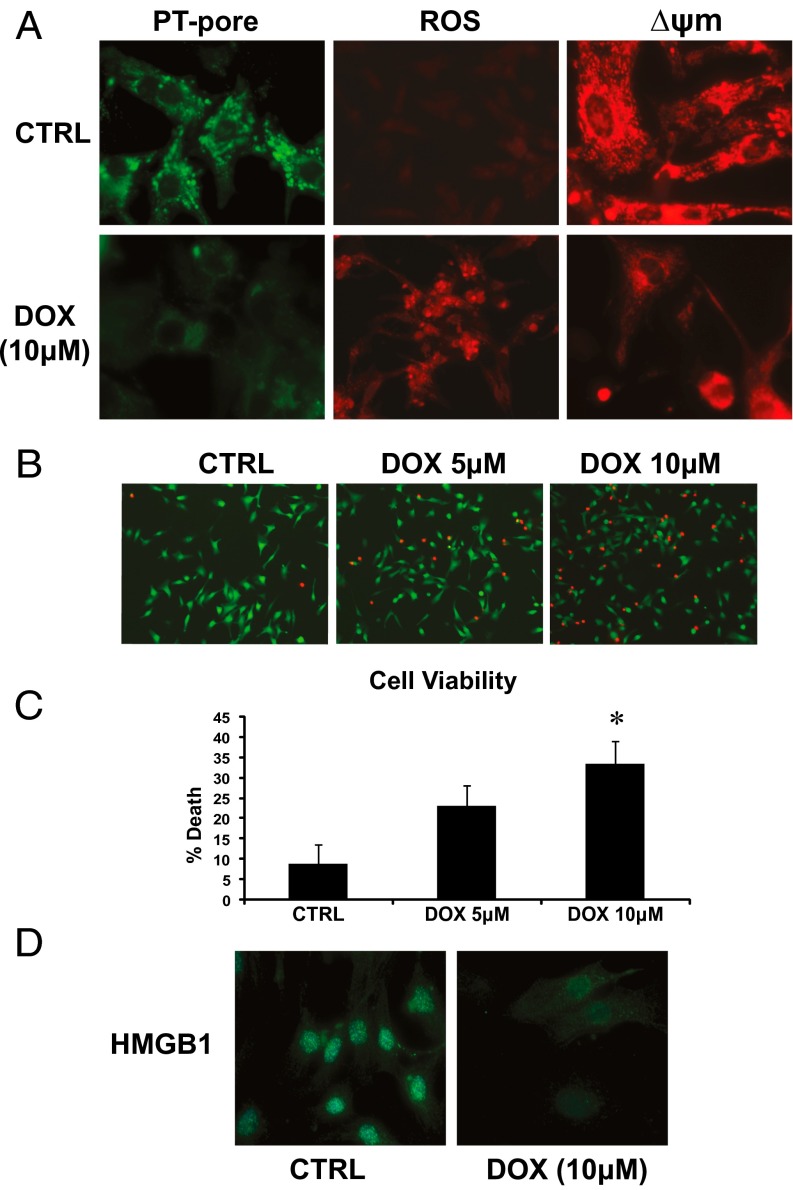
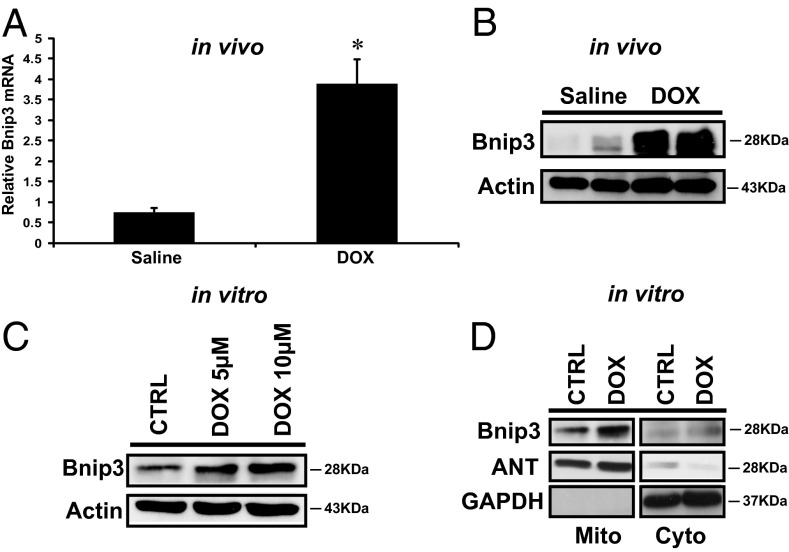
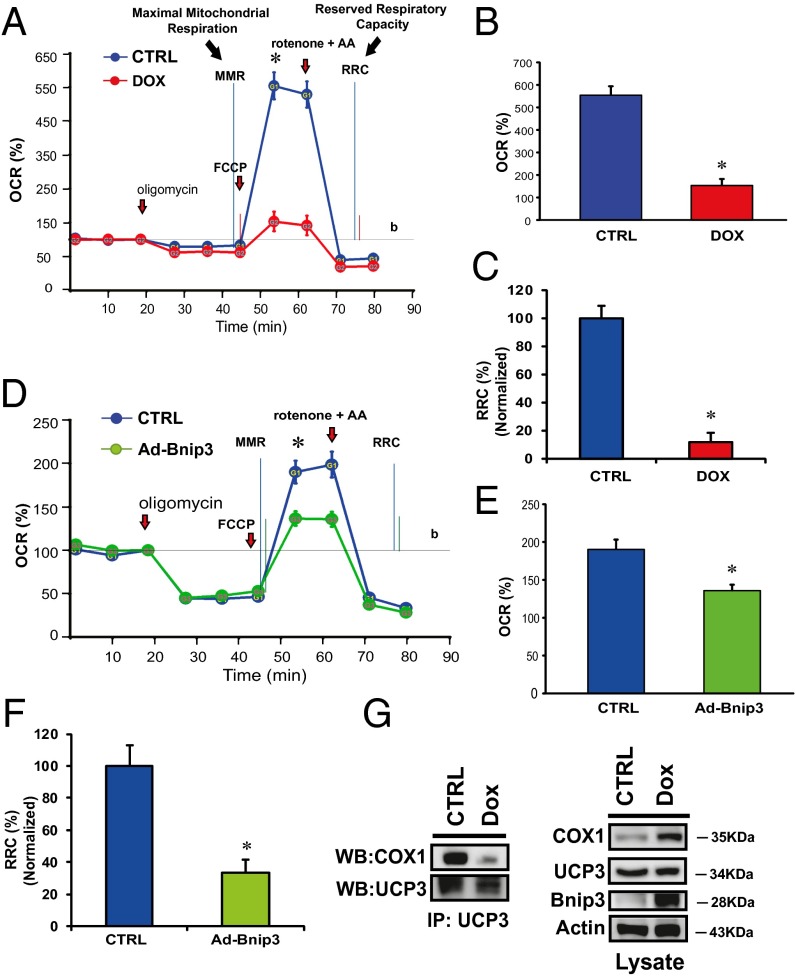
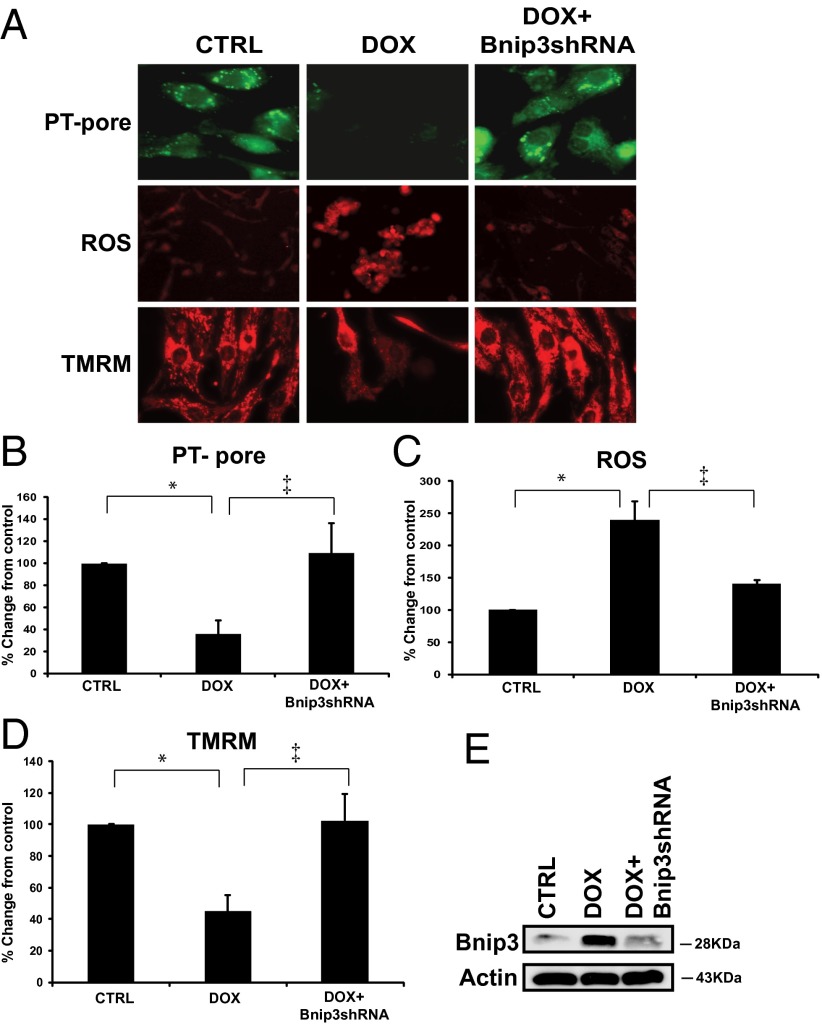
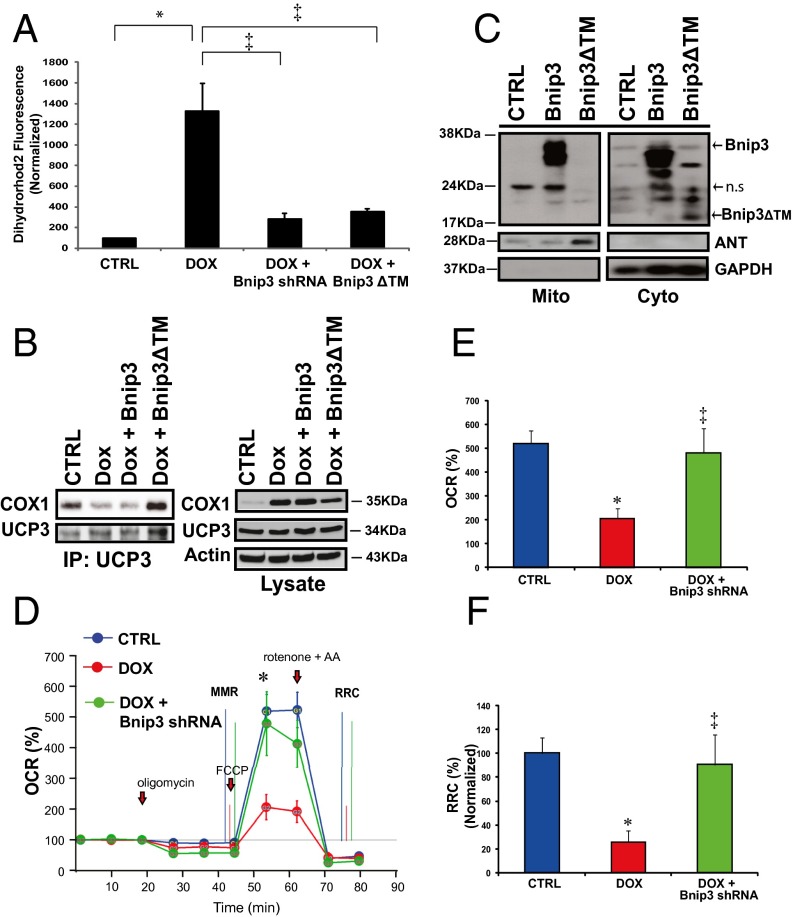
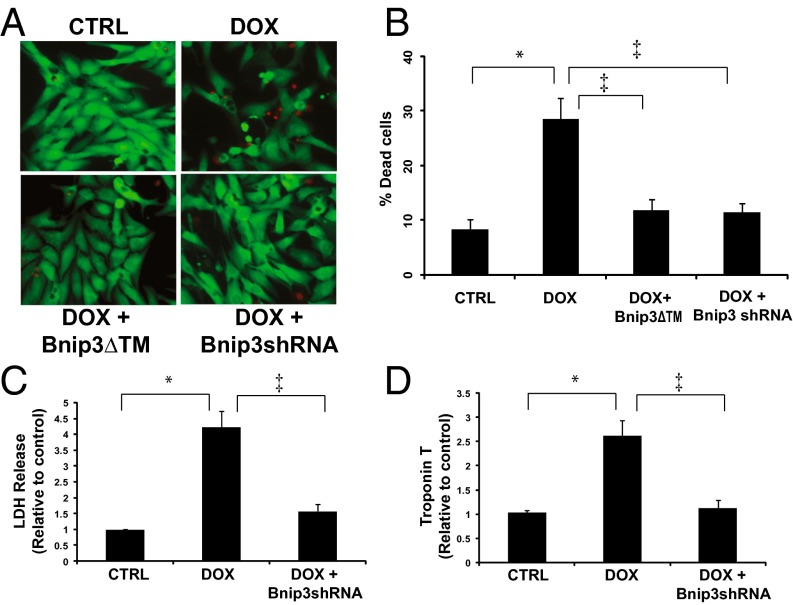
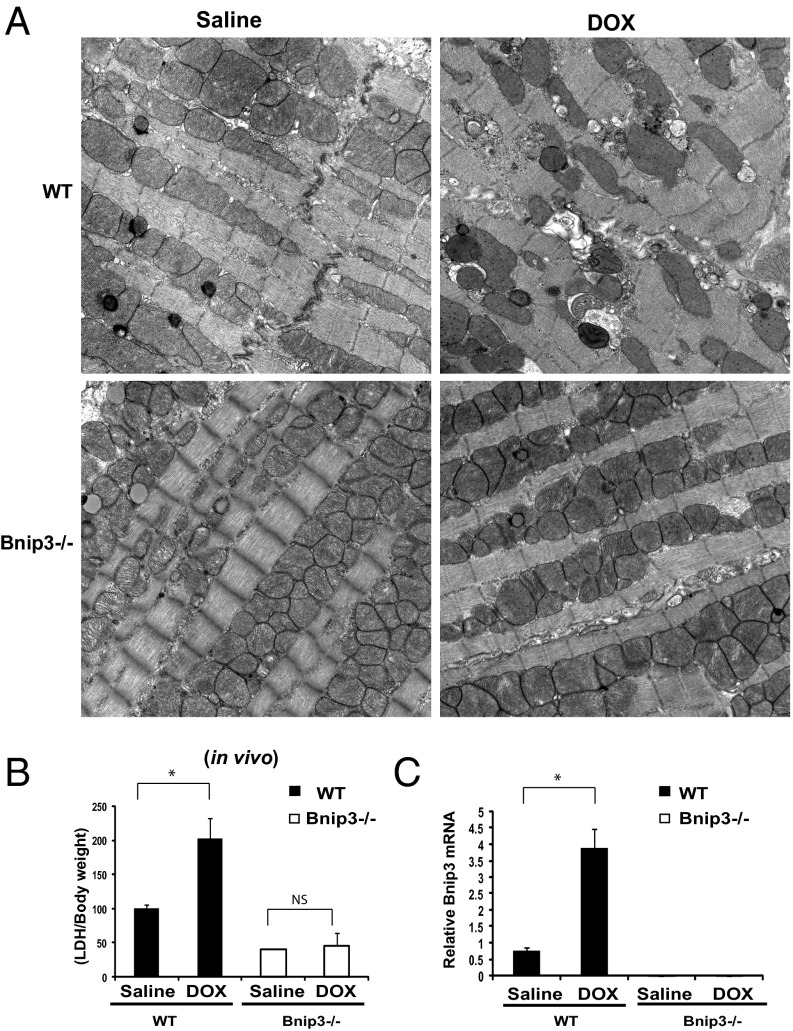
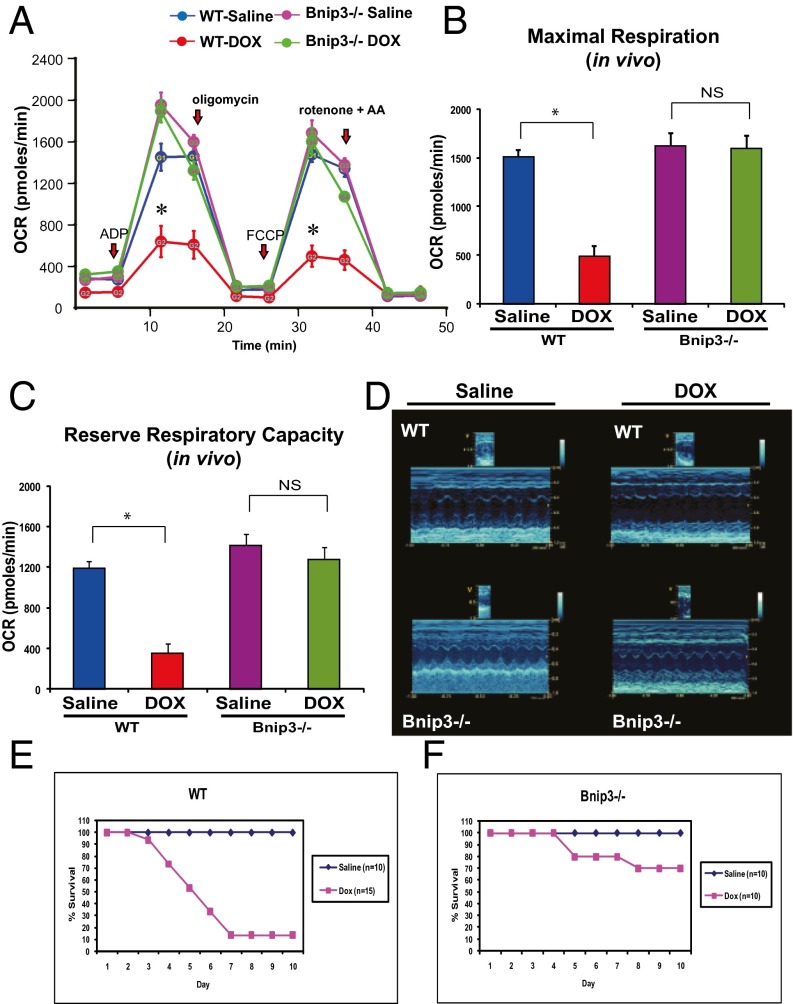
Doxorubicin (DOX) is widely used for treating human cancers, but can induce heart failure through an undefined mechanism. Herein we describe a previously unidentified signaling pathway that couples DOX-induced mitochondrial respiratory chain defects and necrotic cell death to the BH3-only protein Bcl-2-like 19 kDa-interacting protein 3 (Bnip3). Cellular defects, including vacuolization and disrupted mitochondria, were observed in DOX-treated mice hearts. This coincided with mitochondrial localization of Bnip3, increased reactive oxygen species production, loss of mitochondrial membrane potential, mitochondrial permeability transition pore opening, and necrosis. Interestingly, a 3.1-fold decrease in maximal mitochondrial respiration was observed in cardiac mitochondria of mice treated with DOX. In vehicle-treated control cells undergoing normal respiration, the respiratory chain complex IV subunit 1 (COX1) was tightly bound to uncoupling protein 3 (UCP3), but this complex was disrupted in cells treated with DOX. Mitochondrial dysfunction induced by DOX was accompanied by contractile failure and necrotic cell death. Conversely, shRNA directed against Bnip3 or a mutant of Bnip3 defective for mitochondrial targeting abrogated DOX-induced loss of COX1-UCP3 complexes and respiratory chain defects. Finally, Bnip3(-/-) mice treated with DOX displayed relatively normal mitochondrial morphology, respiration, and mortality rates comparable to those of saline-treated WT mice, supporting the idea that Bnip3 underlies the cardiotoxic effects of DOX. These findings reveal a new signaling pathway in which DOX-induced mitochondrial respiratory chain defects and necrotic cell death are mutually dependent on and obligatorily linked to Bnip3 gene activation. Interventions that antagonize Bnip3 may prove beneficial in preventing mitochondrial injury and heart failure in cancer patients undergoing chemotherapy.
Keywords: Bnip3; cell death; heart failure; mitochondria; ventricular myocytes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339(13):900–905. - PubMed
-
- Singal PK, Li T, Kumar D, Danelisen I, Iliskovic N. Adriamycin-induced heart failure: Mechanism and modulation. Mol Cell Biochem. 2000;207(1-2):77–86. - PubMed
-
- Simůnek T, et al. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. 2009;61(1):154–171. - PubMed
-
- Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol. 2007;23(1):15–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
