

Role of Cigarette Smoke-Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis
- PMID: 25490051
- PMCID: PMC5455694
- DOI: 10.1165/rcmb.2014-0107OC
Role of Cigarette Smoke-Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis
Abstract
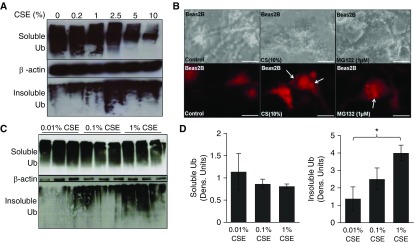
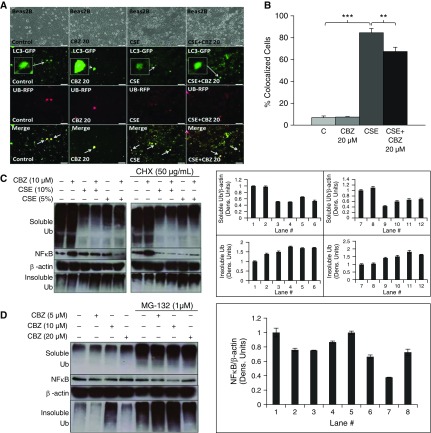
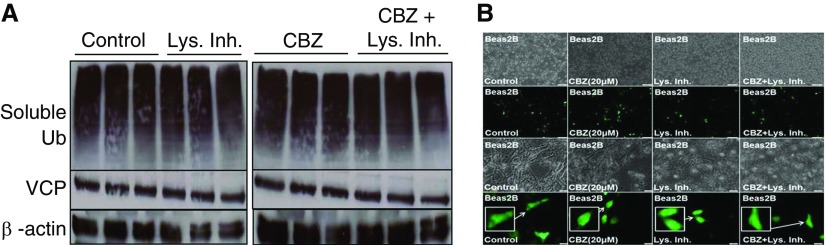
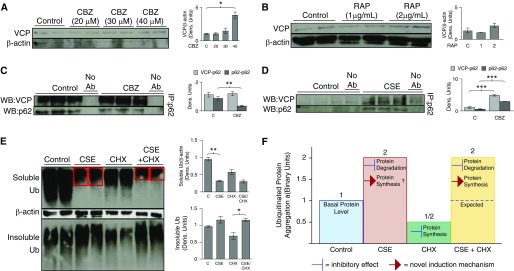
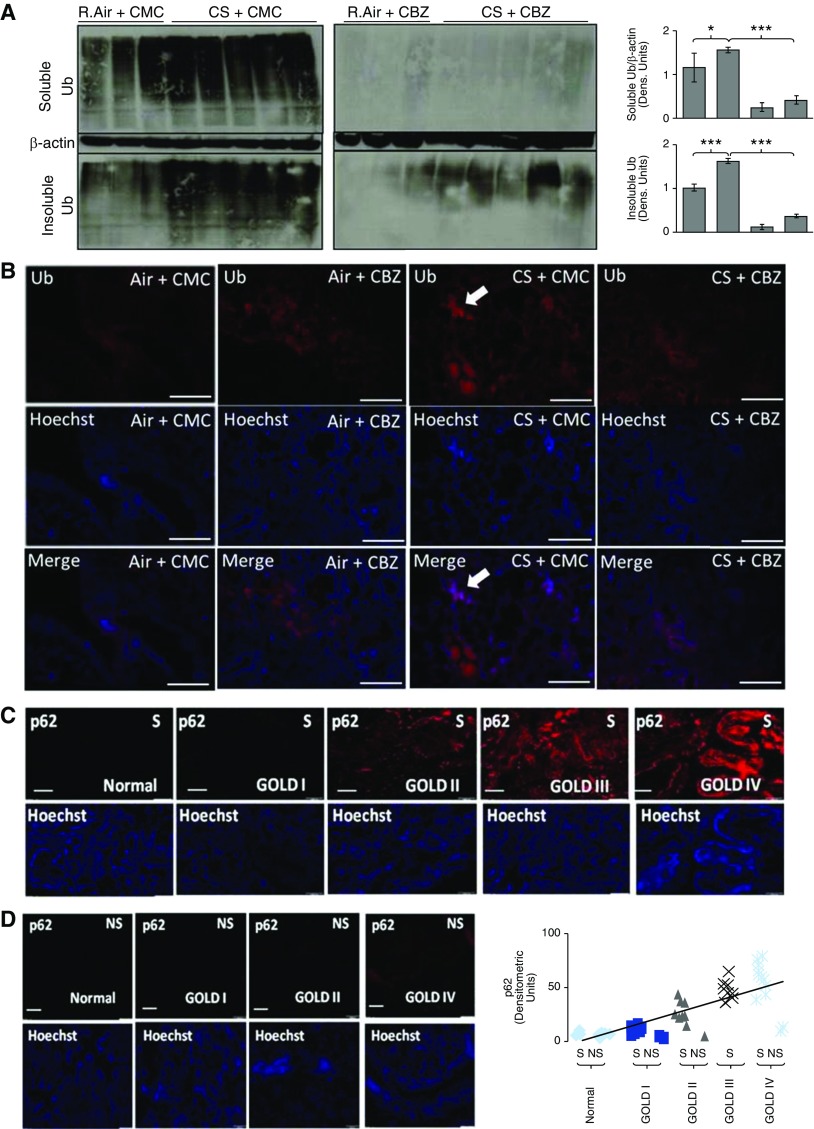
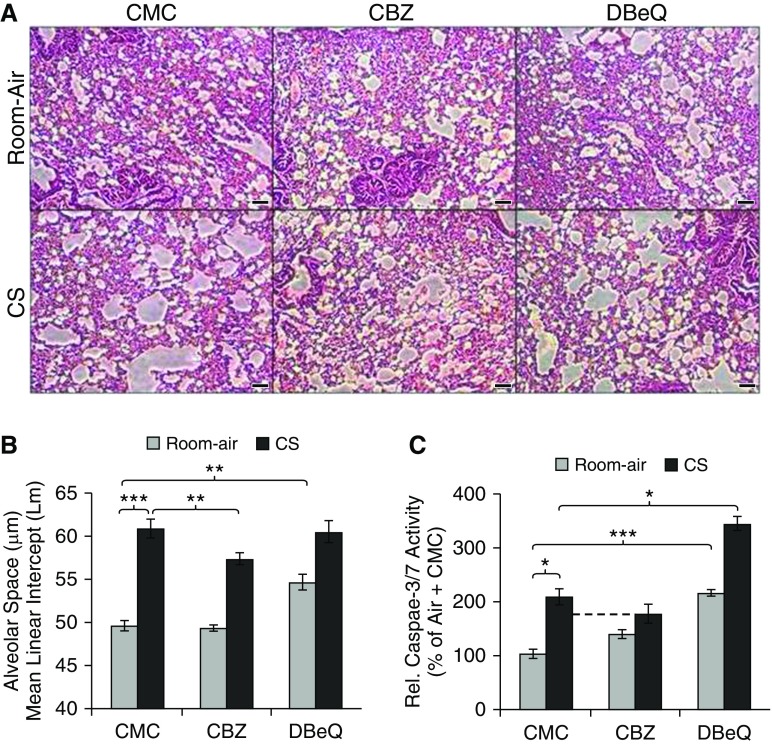
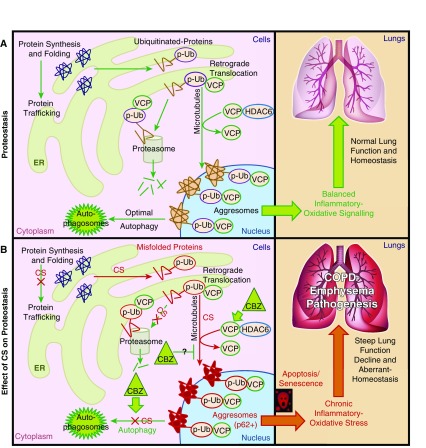
Cigarette smoke (CS) exposure is known to induce proteostasis imbalance that can initiate accumulation of ubiquitinated proteins. Therefore, the primary goal of this study was to determine if first- and secondhand CS induces localization of ubiquitinated proteins in perinuclear spaces as aggresome bodies. Furthermore, we sought to determine the mechanism by which smoke-induced aggresome formation contributes to chronic obstructive pulmonary disease (COPD)-emphysema pathogenesis. Hence, Beas2b cells were treated with CS extract (CSE) for in vitro experimental analysis of CS-induced aggresome formation by immunoblotting, microscopy, and reporter assays, whereas chronic CS-exposed murine model and human COPD-emphysema lung tissues were used for validation. In preliminary analysis, we observed a significant (P < 0.01) increase in ubiquitinated protein aggregation in the insoluble protein fraction of CSE-treated Beas2b cells. We verified that CS-induced ubiquitin aggregrates are localized in the perinuclear spaces as aggresome bodies. These CS-induced aggresomes (P < 0.001) colocalize with autophagy protein microtubule-associated protein 1 light chain-3B(+) autophagy bodies, whereas U.S. Food and Drug Administration-approved autophagy-inducing drug (carbamazepine) significantly (P < 0.01) decreases their colocalization and expression, suggesting CS-impaired autophagy. Moreover, CSE treatment significantly increases valosin-containing protein-p62 protein-protein interaction (P < 0.0005) and p62 expression (aberrant autophagy marker; P < 0.0001), verifying CS-impaired autophagy as an aggresome formation mechanism. We also found that inhibiting protein synthesis by cycloheximide does not deplete CS-induced ubiquitinated protein aggregates, suggesting the role of CS-induced protein synthesis in aggresome formation. Next, we used an emphysema murine model to verify that chronic CS significantly (P < 0.0005) induces aggresome formation. Moreover, we observed that autophagy induction by carbamazepine inhibits CS-induced aggresome formation and alveolar space enlargement (P < 0.001), confirming involvement of aggresome bodies in COPD-emphysema pathogenesis. Finally, significantly higher p62 accumulation in smokers and severe COPD-emphysema lungs (Global Initiative for Chronic Obstructive Lung Disease Stage III/IV) as compared with normal nonsmokers (Global Initiative for Chronic Obstructive Lung Disease Stage 0) substantiates the pathogenic role of autophagy impairment in aggresome formation and COPD-emphysema progression. In conclusion, CS-induced aggresome formation is a novel mechanism involved in COPD-emphysema pathogenesis.
Keywords: aggresomes; autophagy; chronic obstructive pulmonary disease; cigarette smoke; ubiquitin.
Figures
Similar articles
-
Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis.Am J Physiol Cell Physiol. 2018 Jan 1;314(1):C73-C87. doi: 10.1152/ajpcell.00110.2016. Epub 2016 Jul 13. Am J Physiol Cell Physiol. 2018. PMID: 27413169 Free PMC article.
-
Augmentation of S-Nitrosoglutathione Controls Cigarette Smoke-Induced Inflammatory-Oxidative Stress and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis by Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function.Antioxid Redox Signal. 2017 Sep 1;27(7):433-451. doi: 10.1089/ars.2016.6895. Epub 2017 Feb 7. Antioxid Redox Signal. 2017. PMID: 28006950 Free PMC article.
-
Master Autophagy Regulator Transcription Factor EB Regulates Cigarette Smoke-Induced Autophagy Impairment and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis.Antioxid Redox Signal. 2017 Jul 20;27(3):150-167. doi: 10.1089/ars.2016.6842. Epub 2017 Feb 1. Antioxid Redox Signal. 2017. PMID: 27835930 Free PMC article.
-
Augmenting autophagy for prognosis based intervention of COPD-pathophysiology.Respir Res. 2017 May 4;18(1):83. doi: 10.1186/s12931-017-0560-7. Respir Res. 2017. PMID: 28472967 Free PMC article. Review.
-
Nano-based rescue of dysfunctional autophagy in chronic obstructive lung diseases.Expert Opin Drug Deliv. 2017 Apr;14(4):483-489. doi: 10.1080/17425247.2016.1223040. Epub 2016 Aug 26. Expert Opin Drug Deliv. 2017. PMID: 27561233 Review.
Cited by
-
Accumulation of Ubiquitin and Sequestosome-1 Implicate Protein Damage in Diacetyl-Induced Cytotoxicity.Am J Pathol. 2016 Nov;186(11):2887-2908. doi: 10.1016/j.ajpath.2016.07.018. Am J Pathol. 2016. PMID: 27643531 Free PMC article.
-
Therapeutic potential of BLT1 antagonist for COPD: involvement of inducing autophagy and ameliorating inflammation.Drug Des Devel Ther. 2019 Sep 4;13:3105-3116. doi: 10.2147/DDDT.S215433. eCollection 2019. Drug Des Devel Ther. 2019. PMID: 31564828 Free PMC article.
-
E-Liquid Containing a Mixture of Coconut, Vanilla, and Cookie Flavors Causes Cellular Senescence and Dysregulated Repair in Pulmonary Fibroblasts: Implications on Premature Aging.Front Physiol. 2020 Sep 4;11:924. doi: 10.3389/fphys.2020.00924. eCollection 2020. Front Physiol. 2020. PMID: 33013432 Free PMC article.
-
Cigarette smoke promotes COPD by activating platelet-activating factor receptor and inducing neutrophil autophagic death in mice.Oncotarget. 2017 Aug 18;8(43):74720-74735. doi: 10.18632/oncotarget.20353. eCollection 2017 Sep 26. Oncotarget. 2017. PMID: 29088819 Free PMC article.
-
Defining the Neuropathological Aggresome across in Silico, in Vitro, and ex Vivo Experiments.J Phys Chem B. 2021 Mar 4;125(8):1974-1996. doi: 10.1021/acs.jpcb.0c09193. Epub 2021 Jan 19. J Phys Chem B. 2021. PMID: 33464098 Free PMC article. Review.
References
-
- Yoshida T, Tuder RM. Pathobiology of cigarette smoke–induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–1082. - PubMed
-
- Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL, Held LS, Schmid V, Buist S. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27:397–412. - PubMed
-
- Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135:173–180. - PubMed
-
- Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28:219–242. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
