

Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia
- PMID: 25490447
- PMCID: PMC4302418
- DOI: 10.1016/j.ccell.2014.10.012
Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia
Abstract
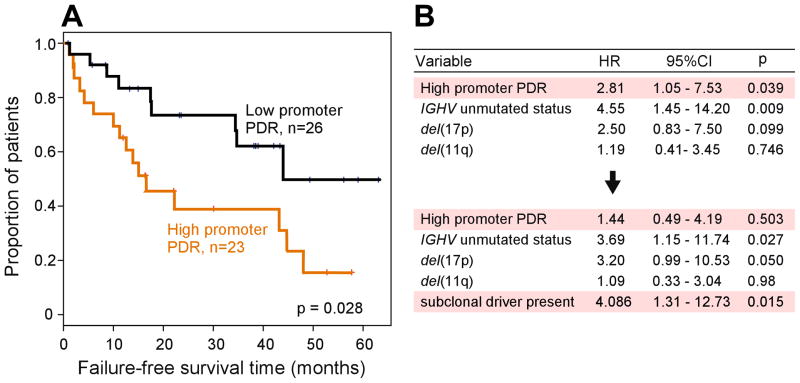
Intratumoral heterogeneity plays a critical role in tumor evolution. To define the contribution of DNA methylation to heterogeneity within tumors, we performed genome-scale bisulfite sequencing of 104 primary chronic lymphocytic leukemias (CLLs). Compared with 26 normal B cell samples, CLLs consistently displayed higher intrasample variability of DNA methylation patterns across the genome, which appears to arise from stochastically disordered methylation in malignant cells. Transcriptome analysis of bulk and single CLL cells revealed that methylation disorder was linked to low-level expression. Disordered methylation was further associated with adverse clinical outcome. We therefore propose that disordered methylation plays a similar role to that of genetic instability, enhancing the ability of cancer cells to search for superior evolutionary trajectories.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures







Comment in
-
Epigenetic noise fuels cancer evolution.Cancer Cell. 2014 Dec 8;26(6):775-776. doi: 10.1016/j.ccell.2014.11.003. Cancer Cell. 2014. PMID: 25490439
References
-
- Akiyama Y, Watkins N, Suzuki H, Jair KW, van Engeland M, Esteller M, Sakai H, Ren CY, Yuasa Y, Herman JG, et al. GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol. 2003;23:8429–8439. - PMC - PubMed
-
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(Suppl 1):S4–11. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- dbGaP/PHS000435.V2.P1
Grants and funding
- 1R01HL103532-01/HL/NHLBI NIH HHS/United States
- R01 CA182461/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- P30 DK043351/DK/NIDDK NIH HHS/United States
- 1K01ES025431-01/ES/NIEHS NIH HHS/United States
- U01ES017155/ES/NIEHS NIH HHS/United States
- 1R01DA036898/DA/NIDA NIH HHS/United States
- R01 HL103532/HL/NHLBI NIH HHS/United States
- U54HG003067/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- K01 ES025431/ES/NIEHS NIH HHS/United States
- P01 CA081534/CA/NCI NIH HHS/United States
- R01 CA184922/CA/NCI NIH HHS/United States
- R01 CA155010/CA/NCI NIH HHS/United States
- 1R01CA155010-01A1/CA/NCI NIH HHS/United States
- U01 ES017155/ES/NIEHS NIH HHS/United States
- R01 DA036898/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
