

An epigenomic roadmap to induced pluripotency reveals DNA methylation as a reprogramming modulator
- PMID: 25493341
- PMCID: PMC4284806
- DOI: 10.1038/ncomms6619
An epigenomic roadmap to induced pluripotency reveals DNA methylation as a reprogramming modulator
Abstract
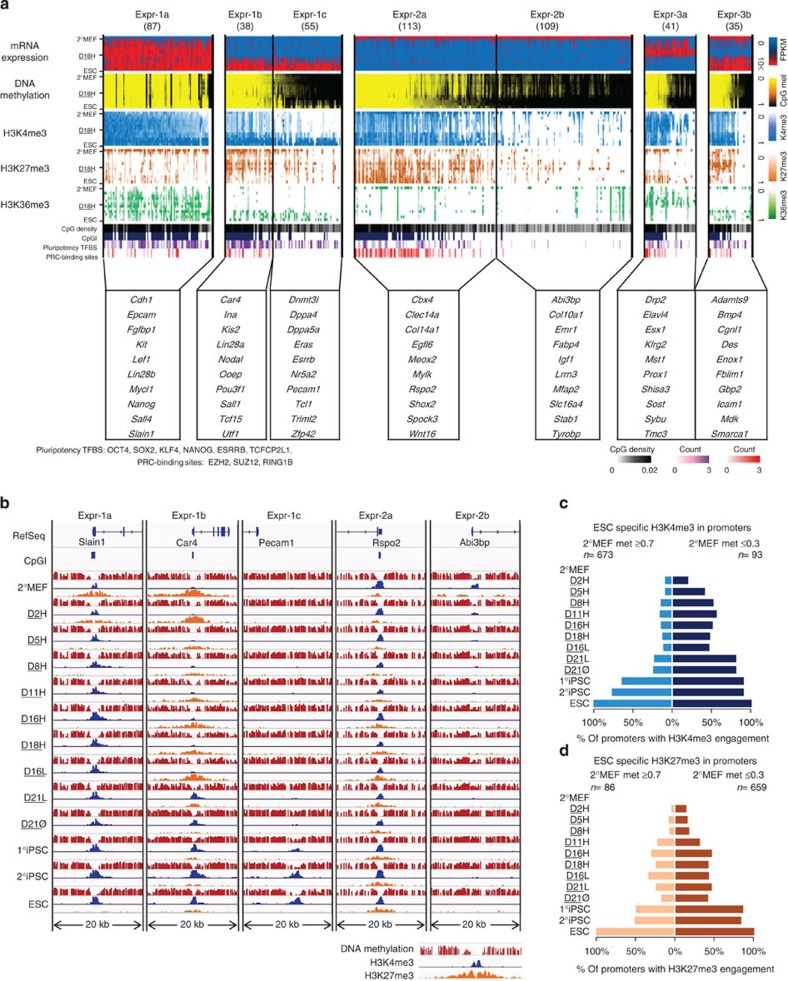
Reprogramming of somatic cells to induced pluripotent stem cells involves a dynamic rearrangement of the epigenetic landscape. To characterize this epigenomic roadmap, we have performed MethylC-seq, ChIP-seq (H3K4/K27/K36me3) and RNA-Seq on samples taken at several time points during murine secondary reprogramming as part of Project Grandiose. We find that DNA methylation gain during reprogramming occurs gradually, while loss is achieved only at the ESC-like state. Binding sites of activated factors exhibit focal demethylation during reprogramming, while ESC-like pluripotent cells are distinguished by extension of demethylation to the wider neighbourhood. We observed that genes with CpG-rich promoters demonstrate stable low methylation and strong engagement of histone marks, whereas genes with CpG-poor promoters are safeguarded by methylation. Such DNA methylation-driven control is the key to the regulation of ESC-pluripotency genes, including Dppa4, Dppa5a and Esrrb. These results reveal the crucial role that DNA methylation plays as an epigenetic switch driving somatic cells to pluripotency.
Figures





References
-
- Takahashi K. & Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). - PubMed
-
- Maherali N. et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1, 55–70 (2007). - PubMed
-
- Takahashi K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). - PubMed
-
- Yu J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007). - PubMed
-
- Park I. H. et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146 (2008). - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
