

Identification of synthetic lethality of PRKDC in MYC-dependent human cancers by pooled shRNA screening
- PMID: 25495526
- PMCID: PMC4320452
- DOI: 10.1186/1471-2407-14-944
Identification of synthetic lethality of PRKDC in MYC-dependent human cancers by pooled shRNA screening
Abstract
Background: MYC family members are among the most frequently deregulated oncogenes in human cancers, yet direct therapeutic targeting of MYC in cancer has been challenging thus far. Synthetic lethality provides an opportunity for therapeutic intervention of MYC-driven cancers.
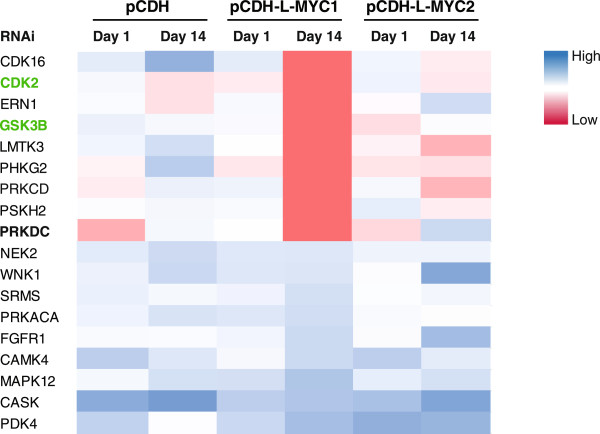
Methods: A pooled kinase shRNA library screen was performed and next-generation deep sequencing efforts identified that PRKDC was synthetically lethal in cells overexpressing MYC. Genes and proteins of interest were knocked down or inhibited using RNAi technology and small molecule inhibitors, respectively. Quantitative RT-PCR using TaqMan probes examined mRNA expression levels and cell viability was assessed using CellTiter-Glo (Promega). Western blotting was performed to monitor different protein levels in the presence or absence of RNAi or compound treatment. Statistical significance of differences among data sets were determined using unpaired t test (Mann-Whitney test) or ANOVA.
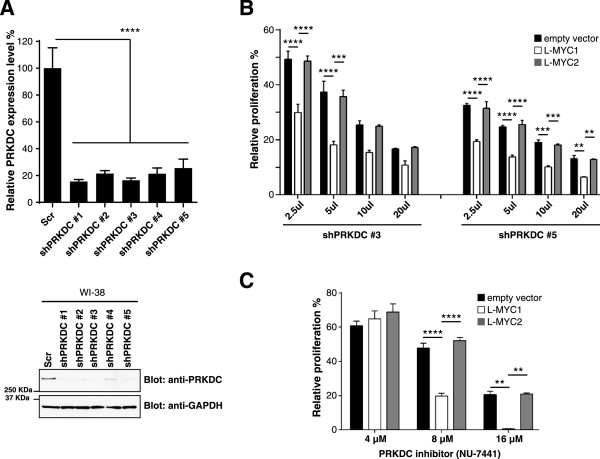
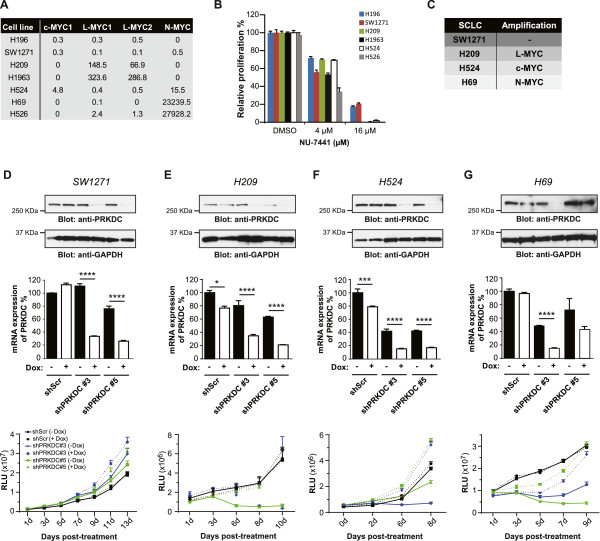
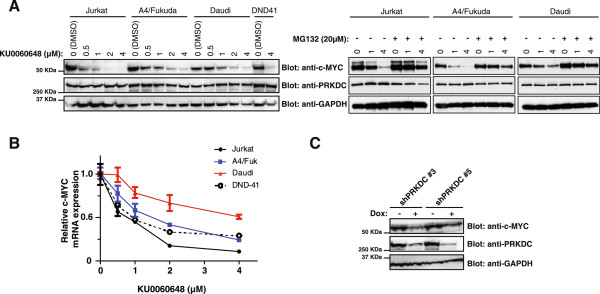
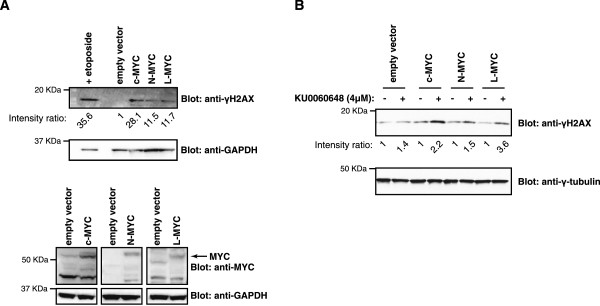
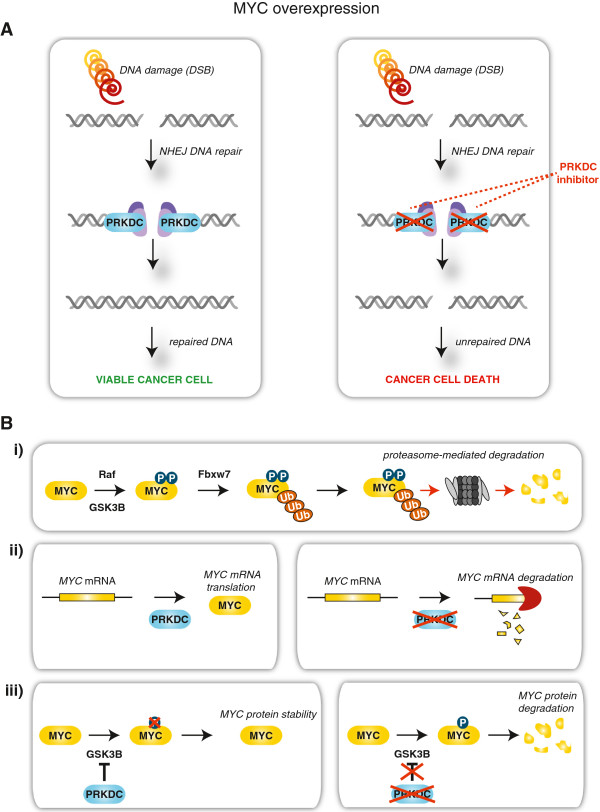
Results: Inhibition of PRKDC using RNAi (RNA interference) or small molecular inhibitors preferentially killed MYC-overexpressing human lung fibroblasts. Moreover, inducible PRKDC knockdown decreased cell viability selectively in high MYC-expressing human small cell lung cancer cell lines. At the molecular level, we found that inhibition of PRKDC downregulated MYC mRNA and protein expression in multiple cancer cell lines. In addition, we confirmed that overexpression of MYC family proteins induced DNA double-strand breaks; our results also revealed that PRKDC inhibition in these cells led to an increase in DNA damage levels.
Conclusions: Our data suggest that the synthetic lethality between PRKDC and MYC may in part be due to PRKDC dependent modulation of MYC expression, as well as MYC-induced DNA damage where PRKDC plays a key role in DNA damage repair.
Figures
References
-
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry K, Pinchback R, Ligon A, Cho Y, Haery L, Greulich H, Reich M, Winckler W, Lawrence M, Weir B, Tanaka K, Chiang D, Bass A, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. - DOI - PMC - PubMed
Pre-publication history
-
- The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/944/prepub
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
