

CLAST: CUDA implemented large-scale alignment search tool
- PMID: 25495907
- PMCID: PMC4271471
- DOI: 10.1186/s12859-014-0406-y
CLAST: CUDA implemented large-scale alignment search tool
Abstract
Background: Metagenomics is a powerful methodology to study microbial communities, but it is highly dependent on nucleotide sequence similarity searching against sequence databases. Metagenomic analyses with next-generation sequencing technologies produce enormous numbers of reads from microbial communities, and many reads are derived from microbes whose genomes have not yet been sequenced, limiting the usefulness of existing sequence similarity search tools. Therefore, there is a clear need for a sequence similarity search tool that can rapidly detect weak similarity in large datasets.
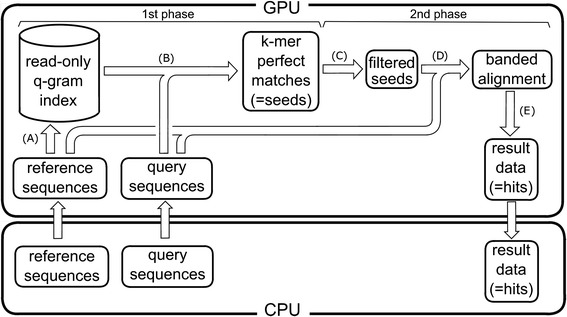
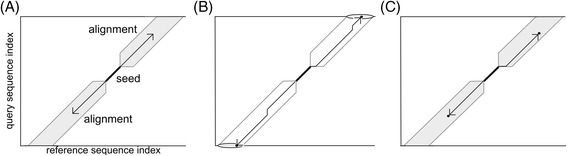
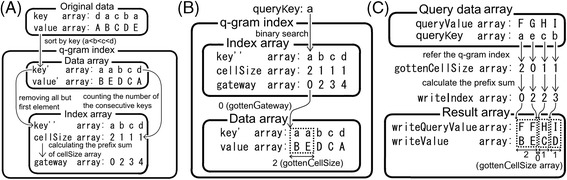
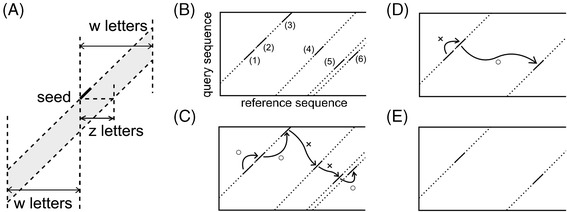
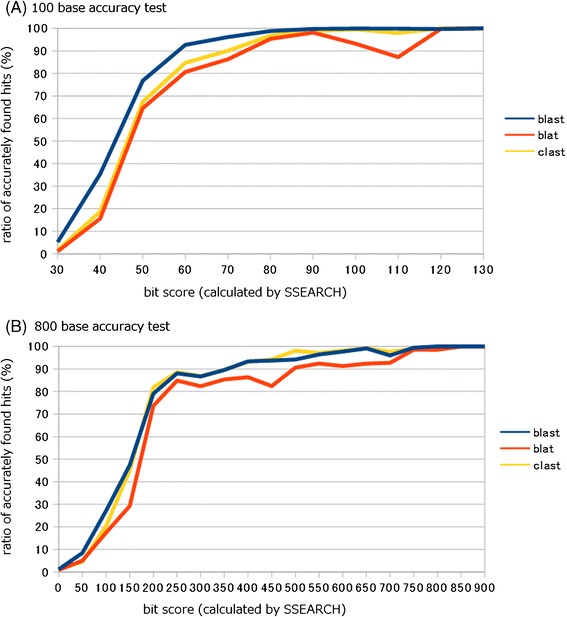
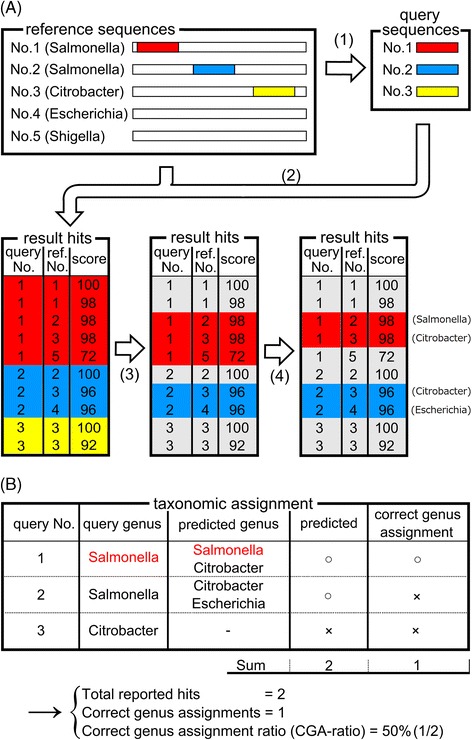
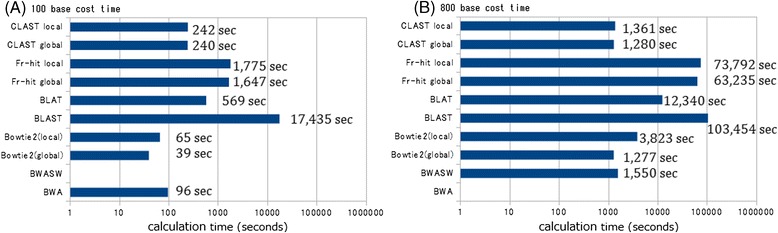
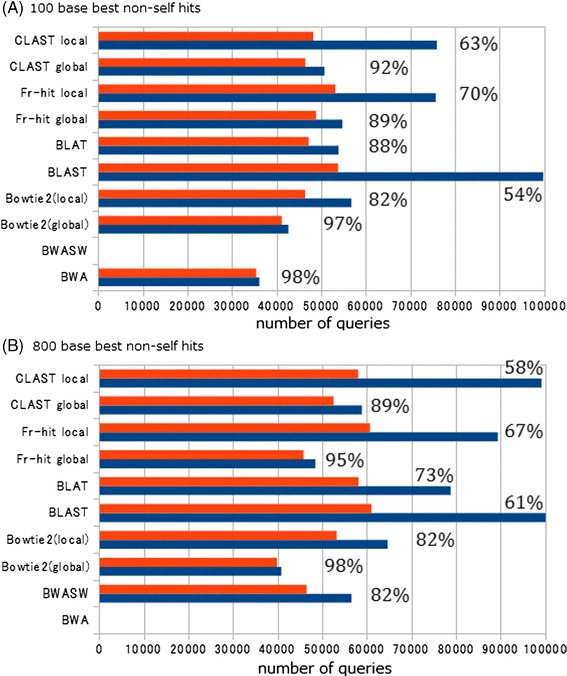
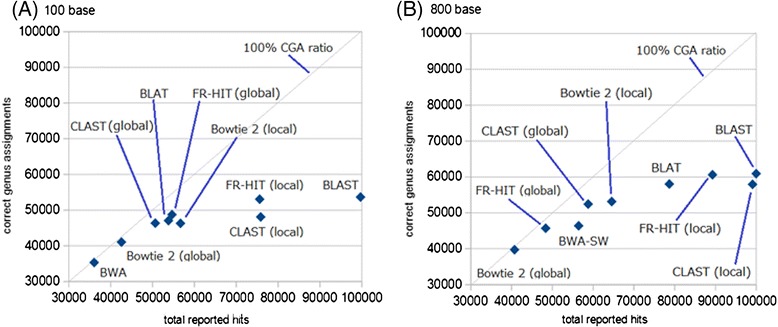
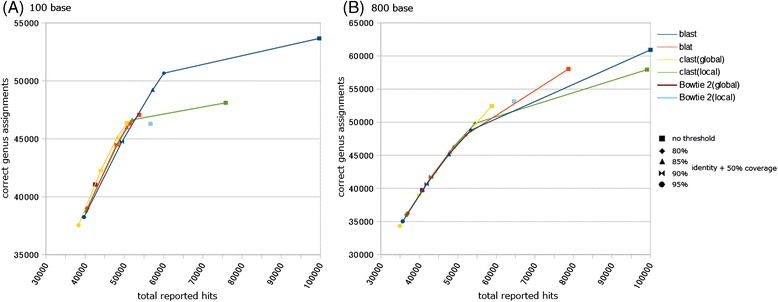
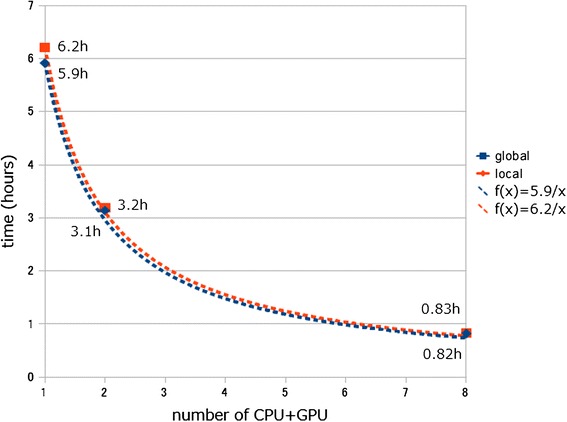
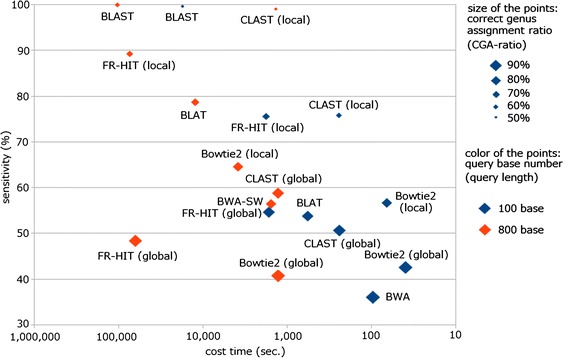
Results: We developed a tool, which we named CLAST (CUDA implemented large-scale alignment search tool), that enables analyses of millions of reads and thousands of reference genome sequences, and runs on NVIDIA Fermi architecture graphics processing units. CLAST has four main advantages over existing alignment tools. First, CLAST was capable of identifying sequence similarities ~80.8 times faster than BLAST and 9.6 times faster than BLAT. Second, CLAST executes global alignment as the default (local alignment is also an option), enabling CLAST to assign reads to taxonomic and functional groups based on evolutionarily distant nucleotide sequences with high accuracy. Third, CLAST does not need a preprocessed sequence database like Burrows-Wheeler Transform-based tools, and this enables CLAST to incorporate large, frequently updated sequence databases. Fourth, CLAST requires <2 GB of main memory, making it possible to run CLAST on a standard desktop computer or server node.
Conclusions: CLAST achieved very high speed (similar to the Burrows-Wheeler Transform-based Bowtie 2 for long reads) and sensitivity (equal to BLAST, BLAT, and FR-HIT) without the need for extensive database preprocessing or a specialized computing platform. Our results demonstrate that CLAST has the potential to be one of the most powerful and realistic approaches to analyze the massive amount of sequence data from next-generation sequencing technologies.
Figures
References
-
- Performance and Specifications for HiSeq 2500/1500 [http://www.illumina.com/systems/hiseq_2500_1500/performance_specificatio...]
-
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
