

High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies
- PMID: 25497548
- PMCID: PMC4272607
- DOI: 10.1016/j.cell.2014.11.015
High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies
Abstract
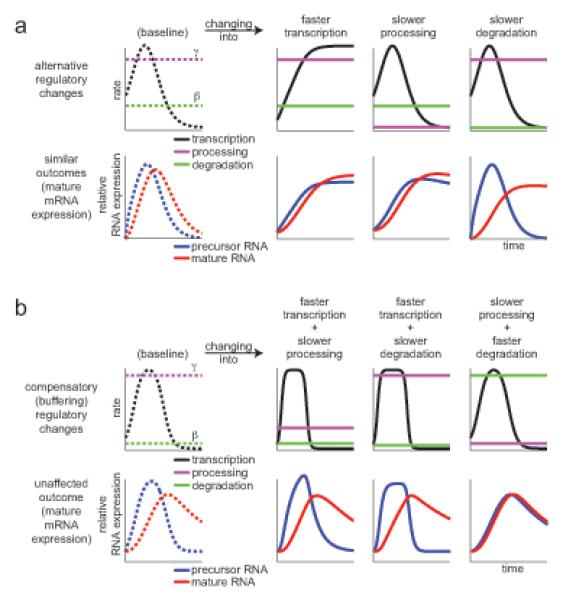
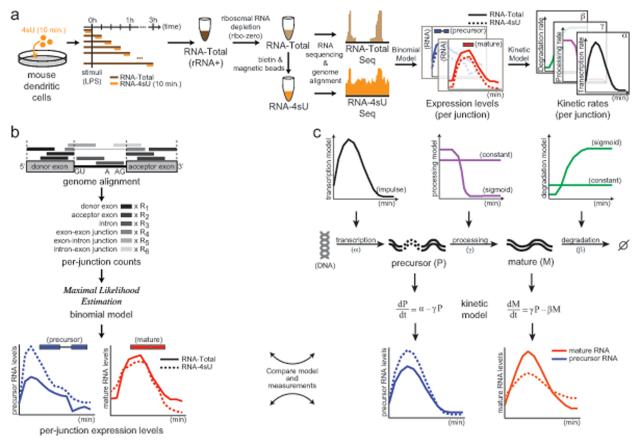
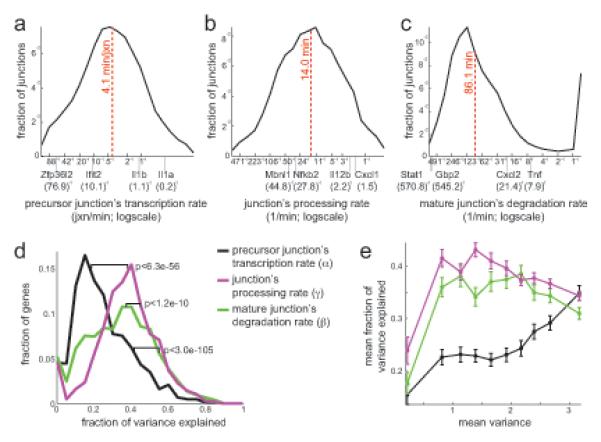
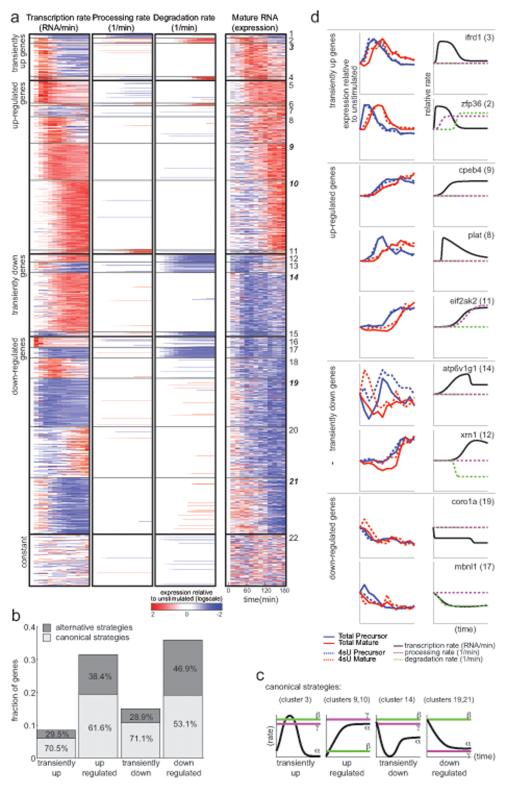
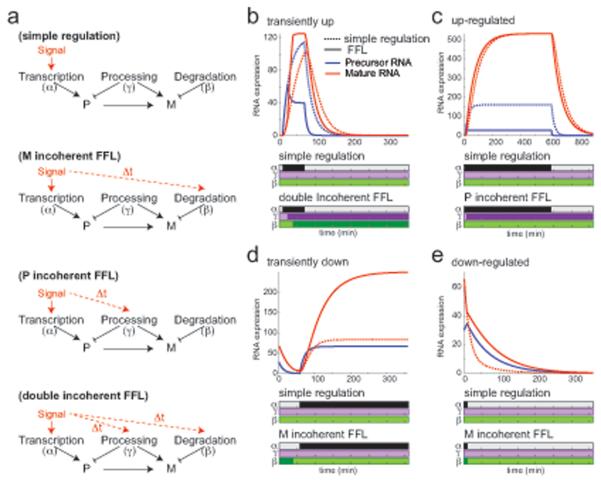
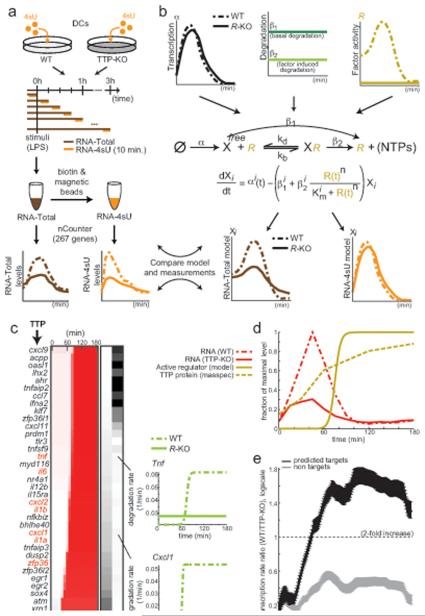
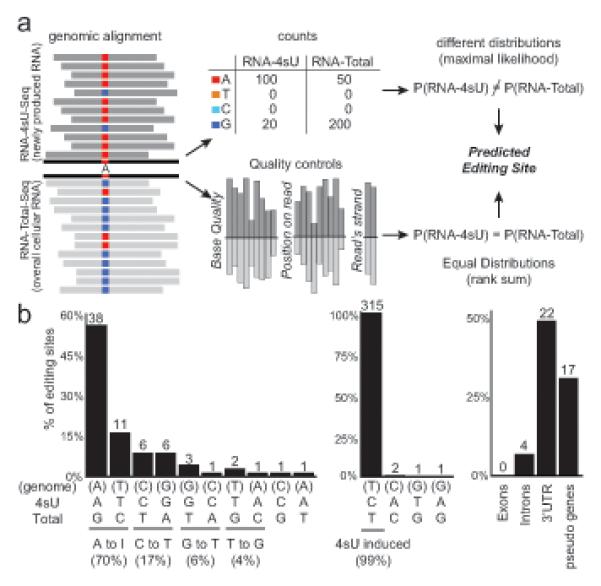
Cells control dynamic transitions in transcript levels by regulating transcription, processing, and/or degradation through an integrated regulatory strategy. Here, we combine RNA metabolic labeling, rRNA-depleted RNA-seq, and DRiLL, a novel computational framework, to quantify the level; editing sites; and transcription, processing, and degradation rates of each transcript at a splice junction resolution during the LPS response of mouse dendritic cells. Four key regulatory strategies, dominated by RNA transcription changes, generate most temporal gene expression patterns. Noncanonical strategies that also employ dynamic posttranscriptional regulation control only a minority of genes, but provide unique signal processing features. We validate Tristetraprolin (TTP) as a major regulator of RNA degradation in one noncanonical strategy. Applying DRiLL to the regulation of noncoding RNAs and to zebrafish embryogenesis demonstrates its broad utility. Our study provides a new quantitative approach to discover transcriptional and posttranscriptional events that control dynamic changes in transcript levels using RNA sequencing data.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
