

Extensive clinical, hormonal and genetic screening in a large consecutive series of 46,XY neonates and infants with atypical sexual development
- PMID: 25497574
- PMCID: PMC4271496
- DOI: 10.1186/s13023-014-0209-2
Extensive clinical, hormonal and genetic screening in a large consecutive series of 46,XY neonates and infants with atypical sexual development
Abstract
Background: One in 4500 children is born with ambiguous genitalia, milder phenotypes occur in one in 300 newborns. Conventional time-consuming hormonal and genetic work-up provides a genetic diagnosis in around 20-40% of 46,XY cases with ambiguous genitalia. All others remain without a definitive diagnosis. The investigation of milder cases, as suggested by recent reports remains controversial.
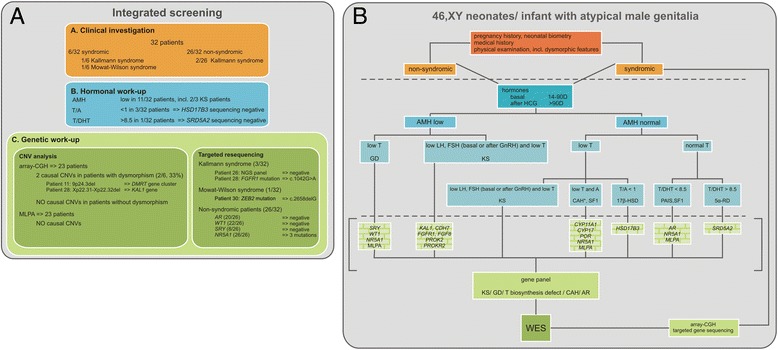
Methods: Integrated clinical, hormonal and genetic screening was performed in a sequential series of 46, XY children, sex-assigned male, who were referred to our pediatric endocrine service for atypical genitalia (2007-2013).
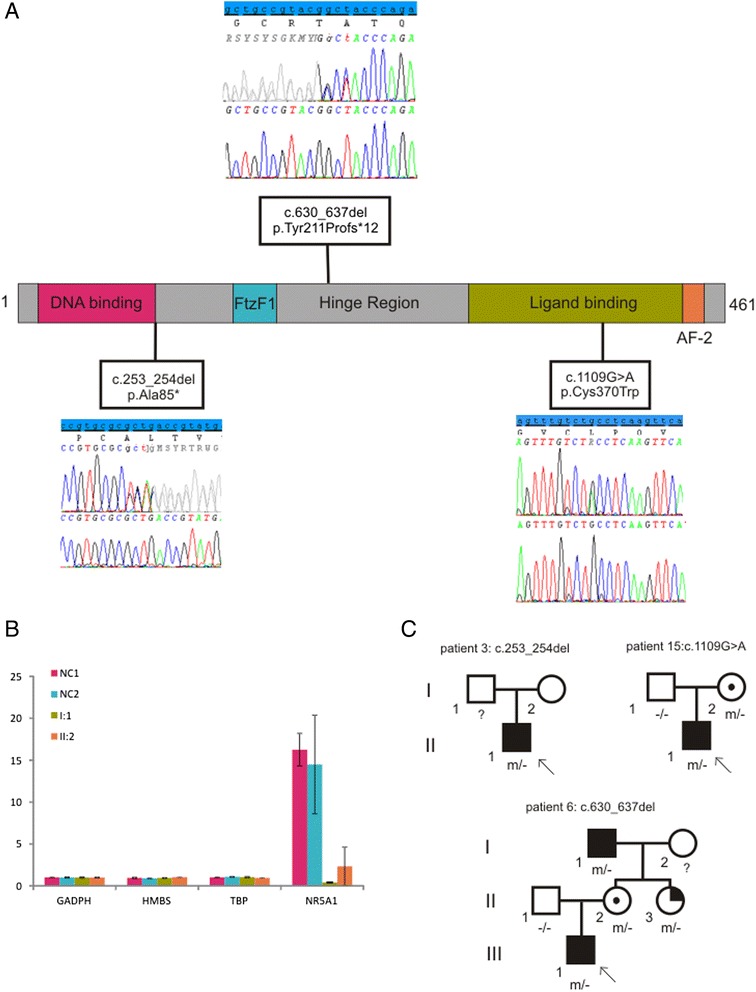
Results: A consecutive cohort of undervirilized 46,XY children with external masculinization score (EMS) 2-12, was extensively investigated. In four patients, a clinical diagnosis of Kallmann syndrome or Mowat-Wilson syndrome was made and genetically supported in 2/3 and 1/1 cases respectively. Hormonal data were suggestive of a (dihydro)testosterone biosynthesis disorder in four cases, however no HSD17B3 or SRD5A2 mutations were found. Array-CGH revealed a causal structural variation in 2/6 syndromic patients. In addition, three novel NR5A1 mutations were found in non-syndromic patients. Interestingly, one mutation was present in a fertile male, underlining the inter- and intrafamilial phenotypic variability of NR5A1-associated phenotypes. No AR, SRY or WT1 mutations were identified.
Conclusion: Overall, a genetic diagnosis could be established in 19% of non-syndromic and 33% of syndromic cases. There is no difference in diagnostic yield between patients with more or less pronounced phenotypes, as expressed by the external masculinisation score (EMS). The clinical utility of array-CGH is high in syndromic cases. Finally, a sequential gene-by-gene approach is time-consuming, expensive and inefficient. Given the low yield and high expense of Sanger sequencing, we anticipate that massively parallel sequencing of gene panels and whole exome sequencing hold promise for genetic diagnosis of 46,XY DSD boys with an undervirilized phenotype.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
