

Novel mutations support a role for Profilin 1 in the pathogenesis of ALS
- PMID: 25499087
- PMCID: PMC4357530
- DOI: 10.1016/j.neurobiolaging.2014.10.032
Novel mutations support a role for Profilin 1 in the pathogenesis of ALS
Abstract
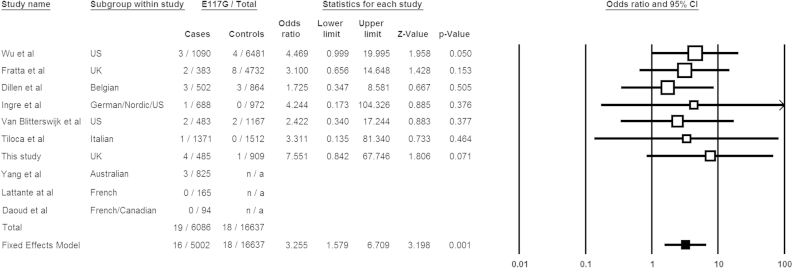
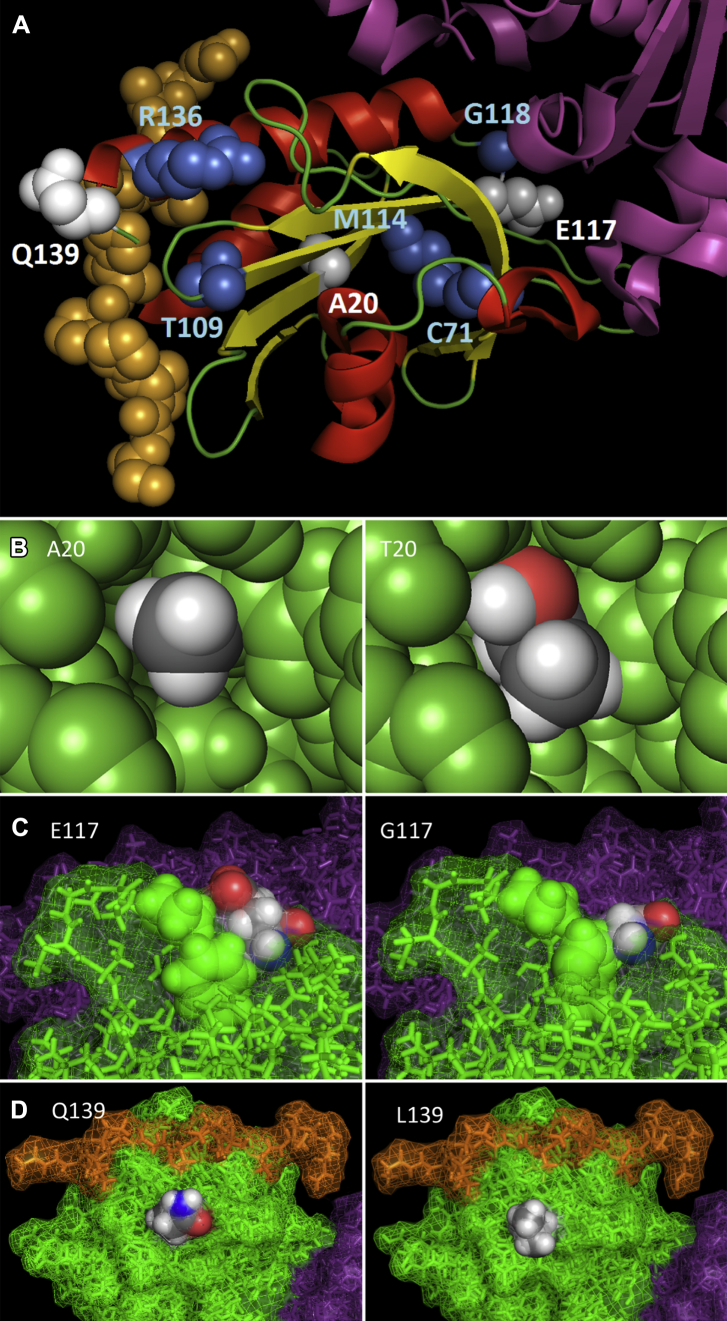
Mutations in the gene encoding profilin 1 (PFN1) have recently been shown to cause amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disorder. We sequenced the PFN1 gene in a cohort of ALS patients (n = 485) and detected 2 novel variants (A20T and Q139L), as well as 4 cases with the previously identified E117G rare variant (∼ 1.2%). A case-control meta-analysis of all published E117G ALS+/- frontotemporal dementia cases including those identified in this report was significant p = 0.001, odds ratio = 3.26 (95% confidence interval, 1.6-6.7), demonstrating this variant to be a susceptibility allele. Postmortem tissue from available patients displayed classic TAR DNA-binding protein 43 pathology. In both transient transfections and in fibroblasts from a patient with the A20T change, we showed that this novel PFN1 mutation causes protein aggregation and the formation of insoluble high molecular weight species which is a hallmark of ALS pathology. Our findings show that PFN1 is a rare cause of ALS and adds further weight to the underlying genetic heterogeneity of this disease.
Keywords: Amyotrophic lateral sclerosis; Profilin 1; TDP-43 proteinopathy.
Crown Copyright © 2015. Published by Elsevier Inc. All rights reserved.
Figures





References
-
- Al-Chalabi A., Jones A., Troakes C., King A., Al-Sarraj S., van den Berg L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012;124:339–352. - PubMed
-
- Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. - PubMed
-
- Brooks B.R. Versailles minimal dataset for diagnosis of ALS: a distillate of the 2nd Consensus Conference on accelerating the diagnosis of ALS. Versailles 2nd Consensus Conference participants. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000;1(Suppl 1):S79–S81. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 089701/WT_/Wellcome Trust/United Kingdom
- DH_/Department of Health/United Kingdom
- 089701/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- G0500289/MRC_/Medical Research Council/United Kingdom
- R01 NS065847/NS/NINDS NIH HHS/United States
- MR/L016397/1/MRC_/Medical Research Council/United Kingdom
- 1R01 NS065847/NS/NINDS NIH HHS/United States
- MR/L021803/1/MRC_/Medical Research Council/United Kingdom
- MC_G1000733/MRC_/Medical Research Council/United Kingdom
- G0900688/MRC_/Medical Research Council/United Kingdom
- MR/L501529/1/MRC_/Medical Research Council/United Kingdom
- R01 NS073873/NS/NINDS NIH HHS/United States
- G1100695/MRC_/Medical Research Council/United Kingdom
- G9318379/MRC_/Medical Research Council/United Kingdom
- MRF-060-0003-RG-SMITH/MRF_/MRF_/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
