

Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes
- PMID: 25501392
- PMCID: PMC4444046
- DOI: 10.1038/ng.3168
Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes
Abstract
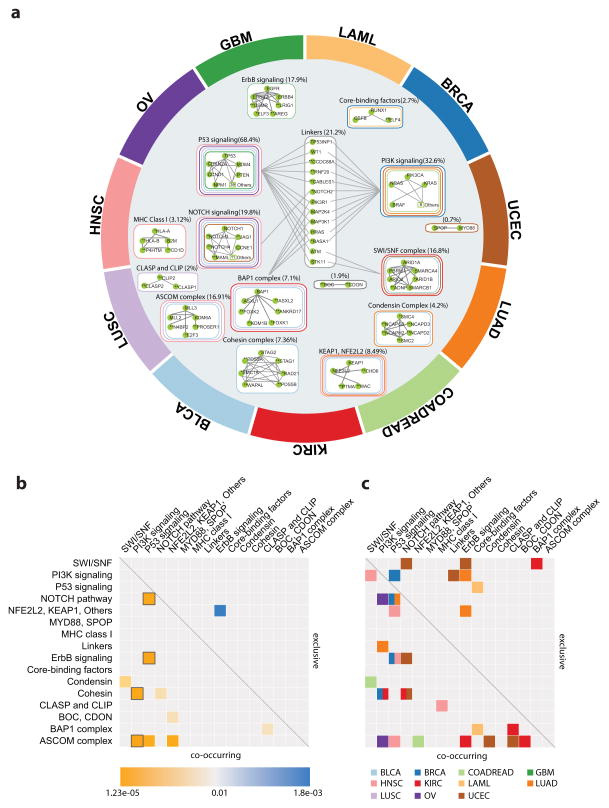
Cancers exhibit extensive mutational heterogeneity, and the resulting long-tail phenomenon complicates the discovery of genes and pathways that are significantly mutated in cancer. We perform a pan-cancer analysis of mutated networks in 3,281 samples from 12 cancer types from The Cancer Genome Atlas (TCGA) using HotNet2, a new algorithm to find mutated subnetworks that overcomes the limitations of existing single-gene, pathway and network approaches. We identify 16 significantly mutated subnetworks that comprise well-known cancer signaling pathways as well as subnetworks with less characterized roles in cancer, including cohesin, condensin and others. Many of these subnetworks exhibit co-occurring mutations across samples. These subnetworks contain dozens of genes with rare somatic mutations across multiple cancers; many of these genes have additional evidence supporting a role in cancer. By illuminating these rare combinations of mutations, pan-cancer network analyses provide a roadmap to investigate new diagnostic and therapeutic opportunities across cancer types.
Conflict of interest statement
A patent application related to this work has been filed.
Figures



Comment in
-
Genetics: HotNet2-see the wood for the trees.Nat Rev Clin Oncol. 2015 Feb;12(2):66. doi: 10.1038/nrclinonc.2014.234. Epub 2015 Jan 6. Nat Rev Clin Oncol. 2015. PMID: 25560530 No abstract available.
-
Co-occurrence of MYC amplification and TP53 mutations in human cancer.Nat Genet. 2016 Feb;48(2):104-6. doi: 10.1038/ng.3468. Nat Genet. 2016. PMID: 26813759 No abstract available.
-
Reply: Co-occurrence of MYC amplification and TP53 mutations in human cancer.Nat Genet. 2016 Feb;48(2):106-8. doi: 10.1038/ng.3491. Nat Genet. 2016. PMID: 26813760 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
