

Genome-wide analysis of DNA methylation dynamics during early human development
- PMID: 25501653
- PMCID: PMC4263407
- DOI: 10.1371/journal.pgen.1004868
Genome-wide analysis of DNA methylation dynamics during early human development
Abstract
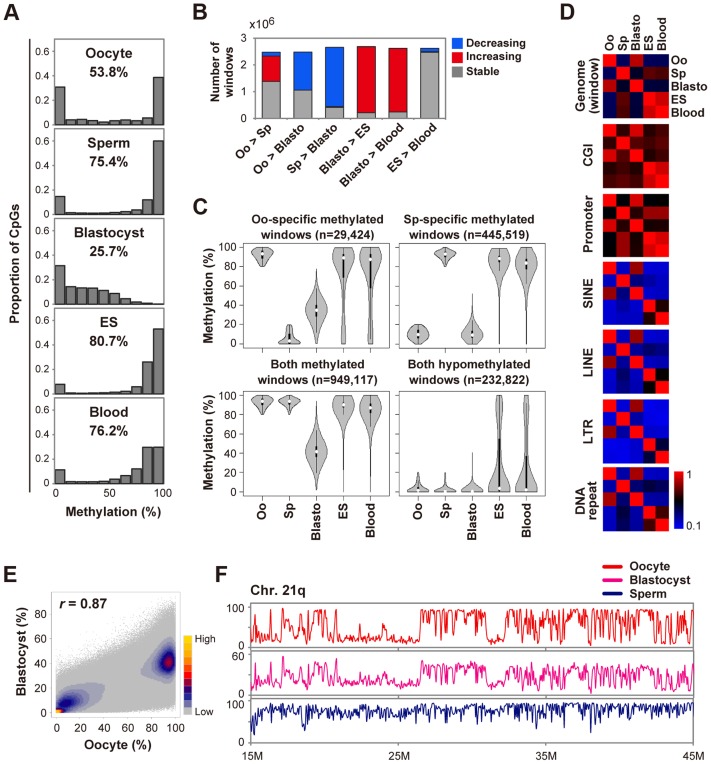
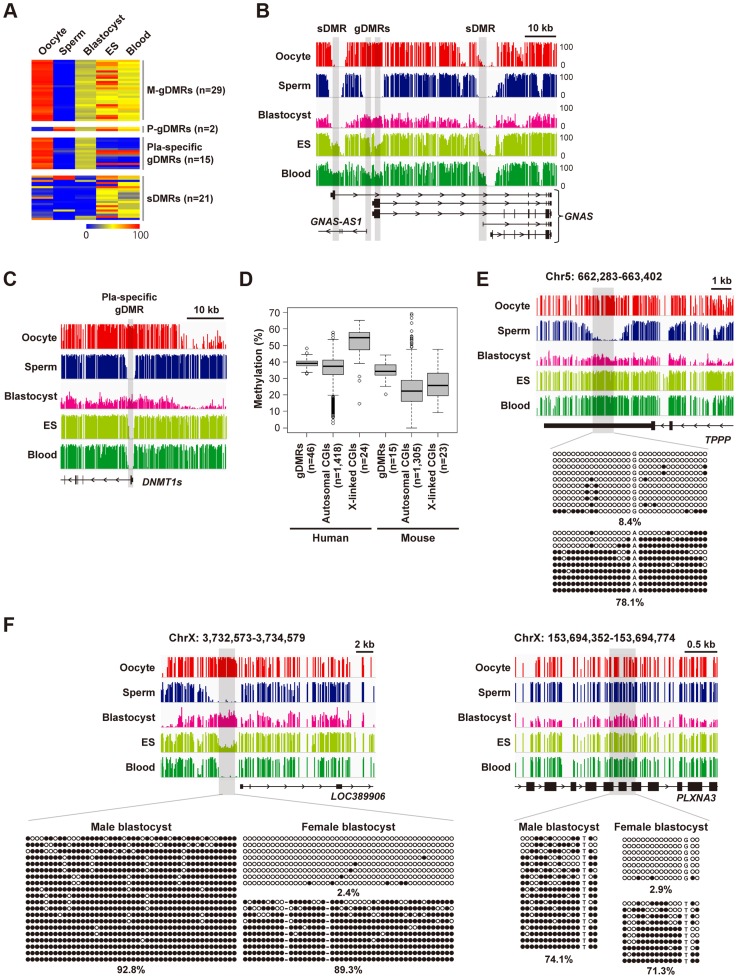
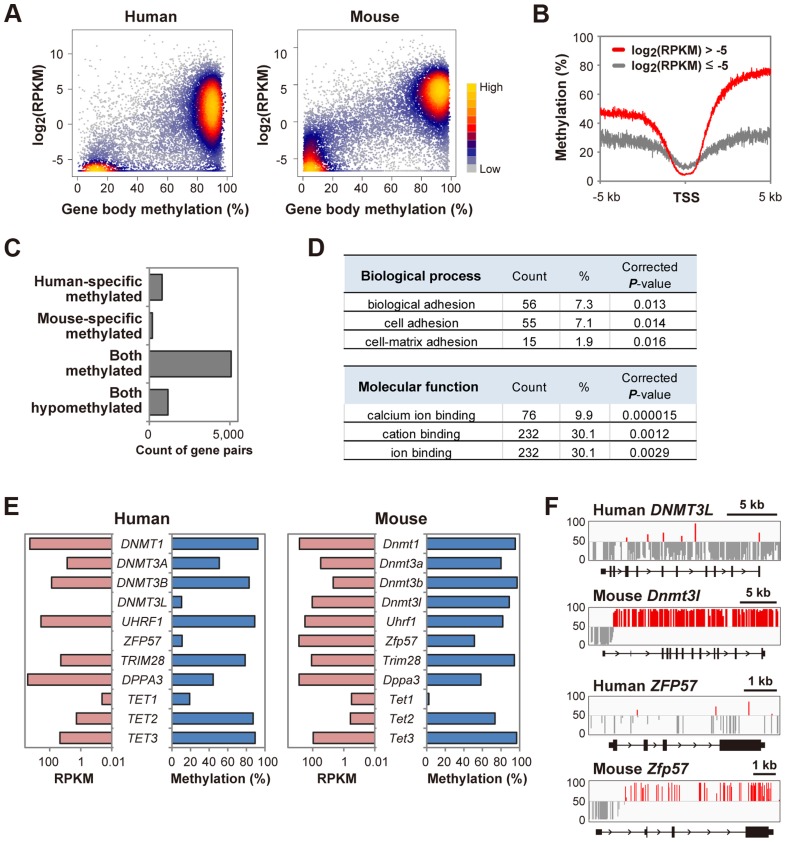
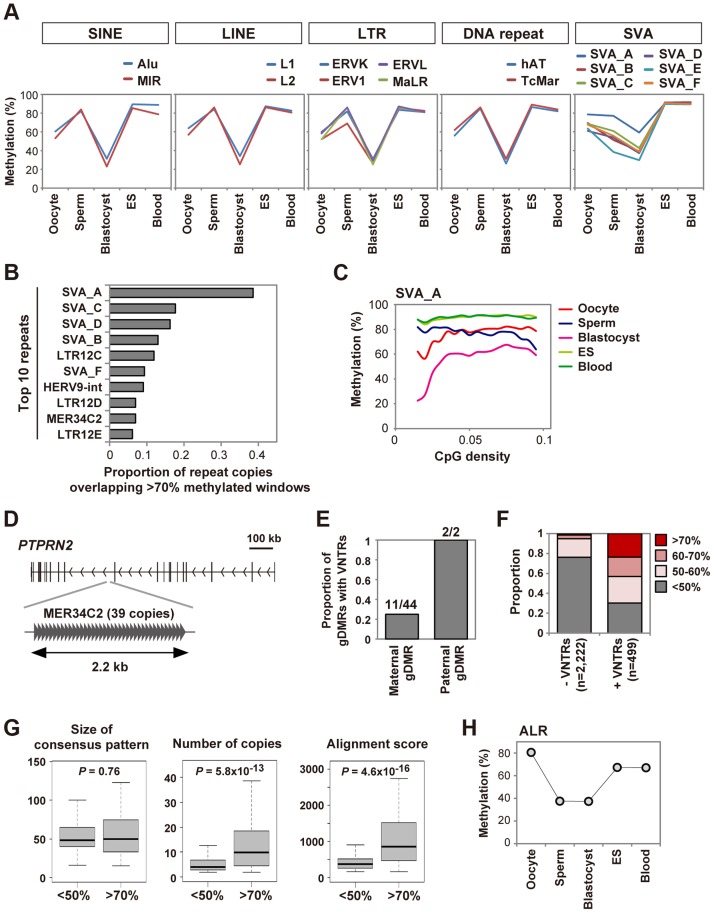
DNA methylation is globally reprogrammed during mammalian preimplantation development, which is critical for normal development. Recent reduced representation bisulfite sequencing (RRBS) studies suggest that the methylome dynamics are essentially conserved between human and mouse early embryos. RRBS is known to cover 5-10% of all genomic CpGs, favoring those contained within CpG-rich regions. To obtain an unbiased and more complete representation of the methylome during early human development, we performed whole genome bisulfite sequencing of human gametes and blastocysts that covered>70% of all genomic CpGs. We found that the maternal genome was demethylated to a much lesser extent in human blastocysts than in mouse blastocysts, which could contribute to an increased number of imprinted differentially methylated regions in the human genome. Global demethylation of the paternal genome was confirmed, but SINE-VNTR-Alu elements and some other tandem repeat-containing regions were found to be specifically protected from this global demethylation. Furthermore, centromeric satellite repeats were hypermethylated in human oocytes but not in mouse oocytes, which might be explained by differential expression of de novo DNA methyltransferases. These data highlight both conserved and species-specific regulation of DNA methylation during early mammalian development. Our work provides further information critical for understanding the epigenetic processes underlying differentiation and pluripotency during early human development.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
