

Identification of rare causal variants in sequence-based studies: methods and applications to VPS13B, a gene involved in Cohen syndrome and autism
- PMID: 25502226
- PMCID: PMC4263785
- DOI: 10.1371/journal.pgen.1004729
Identification of rare causal variants in sequence-based studies: methods and applications to VPS13B, a gene involved in Cohen syndrome and autism
Abstract
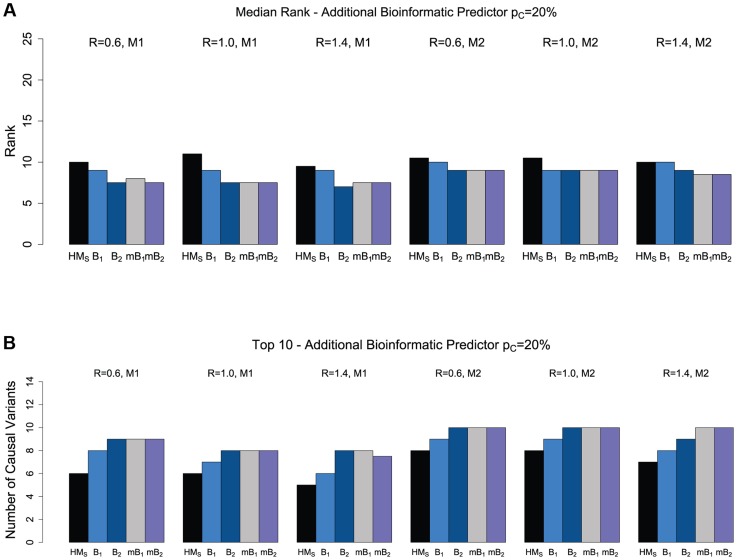
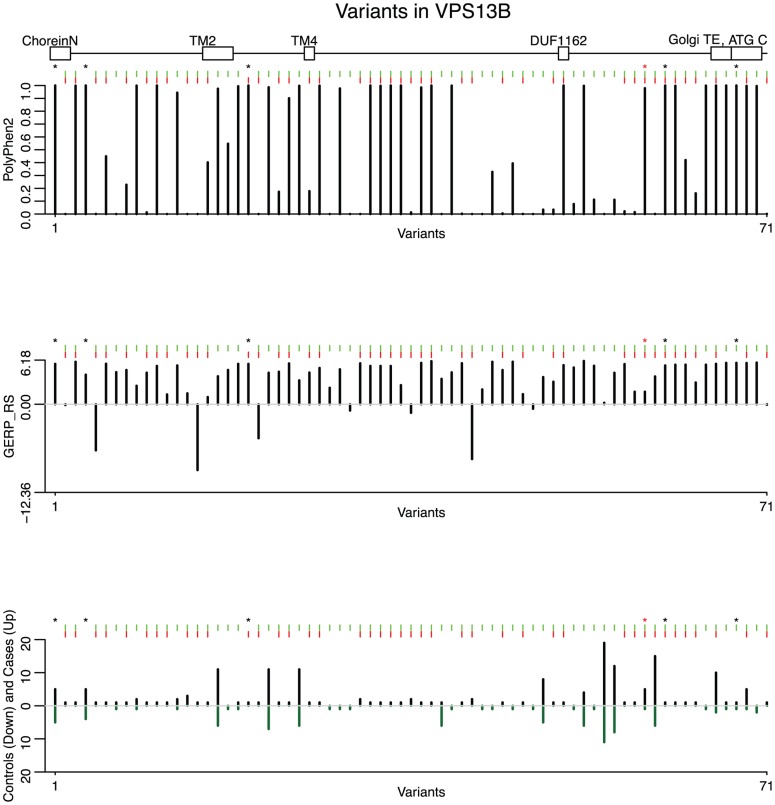
Pinpointing the small number of causal variants among the abundant naturally occurring genetic variation is a difficult challenge, but a crucial one for understanding precise molecular mechanisms of disease and follow-up functional studies. We propose and investigate two complementary statistical approaches for identification of rare causal variants in sequencing studies: a backward elimination procedure based on groupwise association tests, and a hierarchical approach that can integrate sequencing data with diverse functional and evolutionary conservation annotations for individual variants. Using simulations, we show that incorporation of multiple bioinformatic predictors of deleteriousness, such as PolyPhen-2, SIFT and GERP++ scores, can improve the power to discover truly causal variants. As proof of principle, we apply the proposed methods to VPS13B, a gene mutated in the rare neurodevelopmental disorder called Cohen syndrome, and recently reported with recessive variants in autism. We identify a small set of promising candidates for causal variants, including two loss-of-function variants and a rare, homozygous probably-damaging variant that could contribute to autism risk.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


References
-
- Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24: 133–141. - PubMed
-
- Metzker ML (2010) Sequencing technologies - the next generation. Nat Rev Genet 11: 31–46. - PubMed
-
- Ionita-Laza I, Cho MH, Laird NM (2013) Statistical Challenges in Sequence-Based Association Studies with Population-and Family-Based Designs. Statistics in Biosciences 5: 54–70.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
