

Characterization of X chromosome inactivation using integrated analysis of whole-exome and mRNA sequencing
- PMID: 25503791
- PMCID: PMC4264736
- DOI: 10.1371/journal.pone.0113036
Characterization of X chromosome inactivation using integrated analysis of whole-exome and mRNA sequencing
Abstract
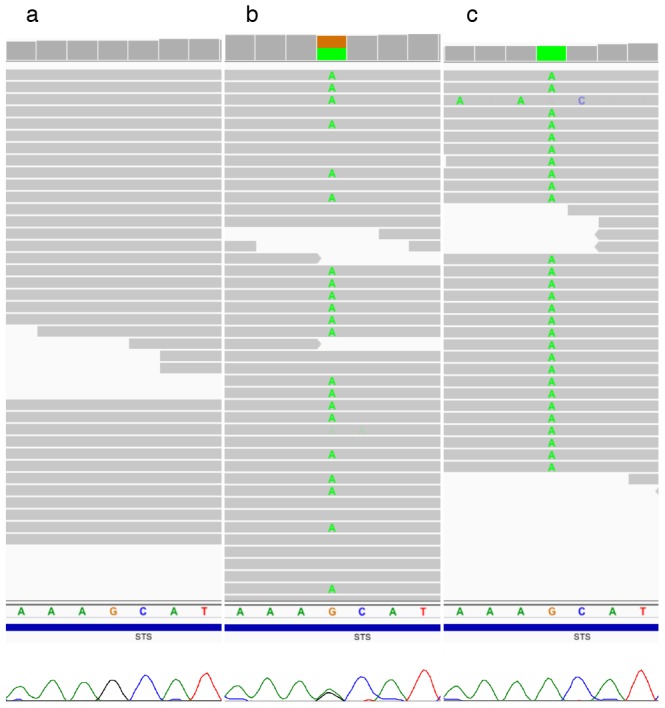
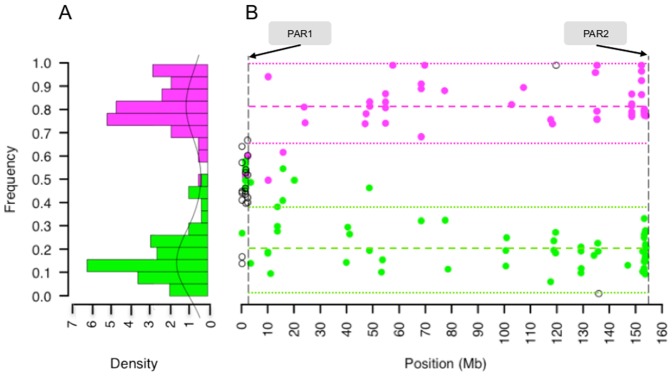
In females, X chromosome inactivation (XCI) is an epigenetic, gene dosage compensatory mechanism by inactivation of one copy of X in cells. Random XCI of one of the parental chromosomes results in an approximately equal proportion of cells expressing alleles from either the maternally or paternally inherited active X, and is defined by the XCI ratio. Skewed XCI ratio is suggestive of non-random inactivation, which can play an important role in X-linked genetic conditions. Current methods rely on indirect, semi-quantitative DNA methylation-based assay to estimate XCI ratio. Here we report a direct approach to estimate XCI ratio by integrated, family-trio based whole-exome and mRNA sequencing using phase-by-transmission of alleles coupled with allele-specific expression analysis. We applied this method to in silico data and to a clinical patient with mild cognitive impairment but no clear diagnosis or understanding molecular mechanism underlying the phenotype. Simulation showed that phased and unphased heterozygous allele expression can be used to estimate XCI ratio. Segregation analysis of the patient's exome uncovered a de novo, interstitial, 1.7 Mb deletion on Xp22.31 that originated on the paternally inherited X and previously been associated with heterogeneous, neurological phenotype. Phased, allelic expression data suggested an 83∶20 moderately skewed XCI that favored the expression of the maternally inherited, cytogenetically normal X and suggested that the deleterious affect of the de novo event on the paternal copy may be offset by skewed XCI that favors expression of the wild-type X. This study shows the utility of integrated sequencing approach in XCI ratio estimation.
Conflict of interest statement
Figures
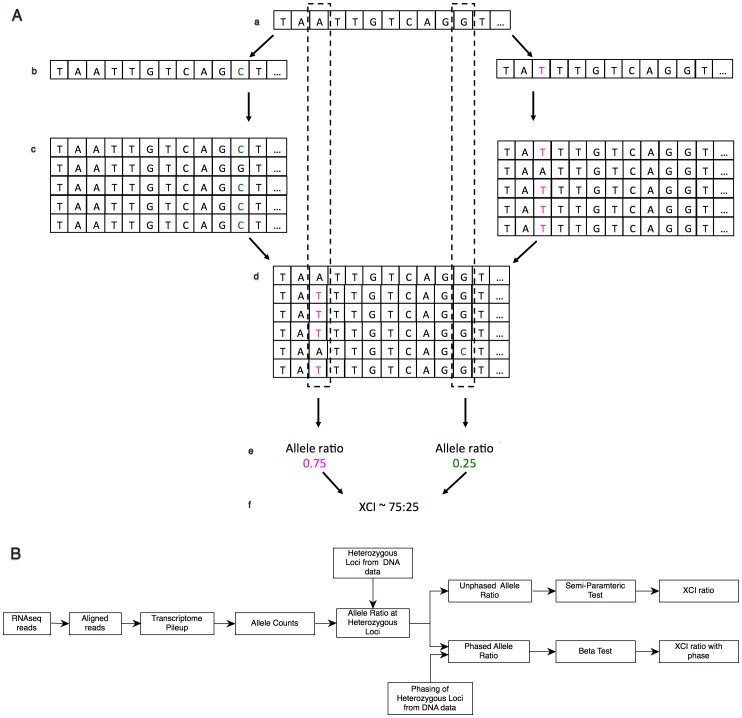
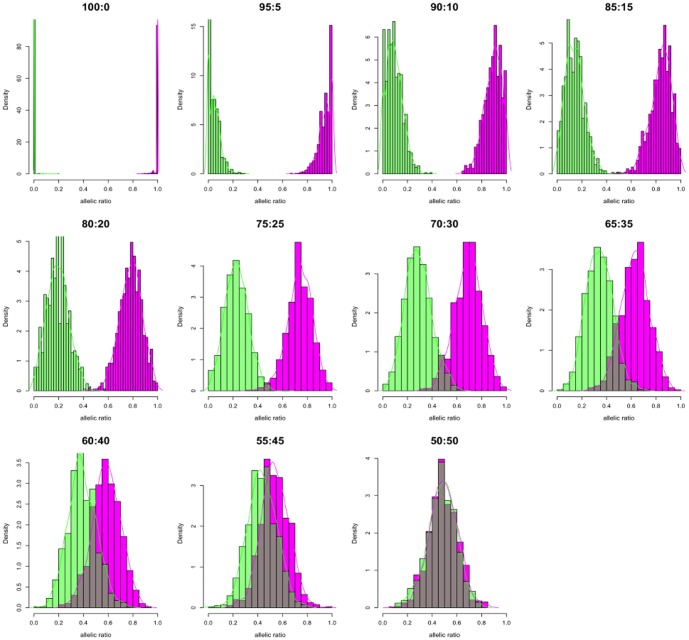
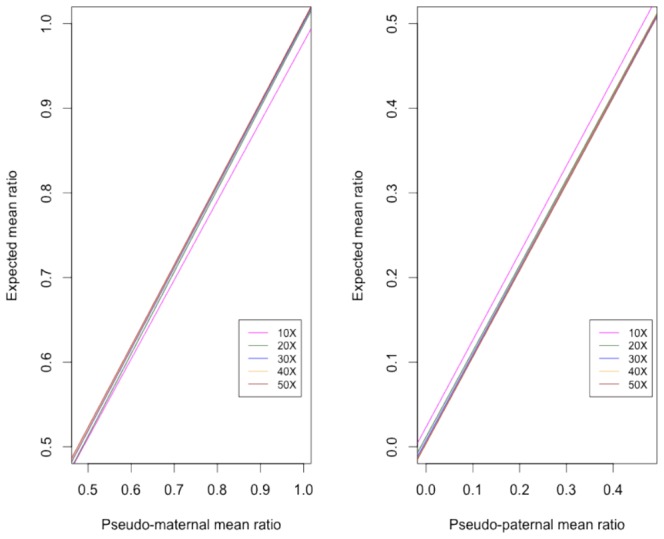
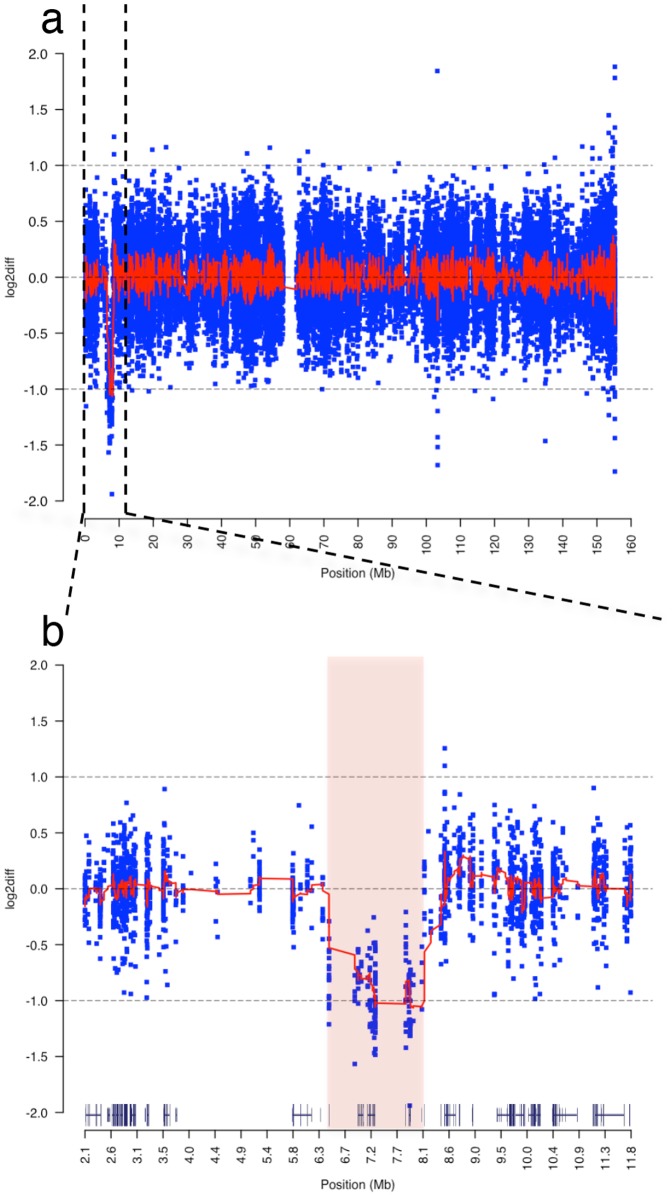
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
