

Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently
- PMID: 25503938
- PMCID: PMC4349129
- DOI: 10.1039/c4cs00351a
Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently
Abstract
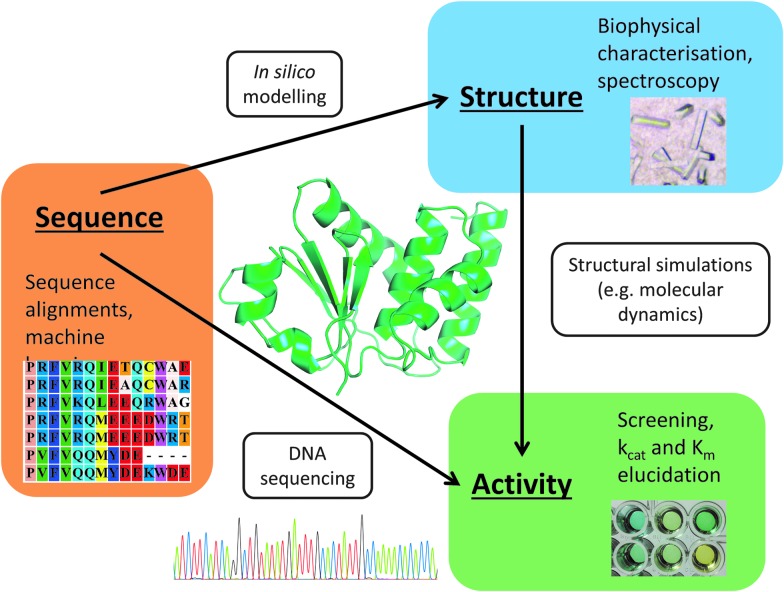
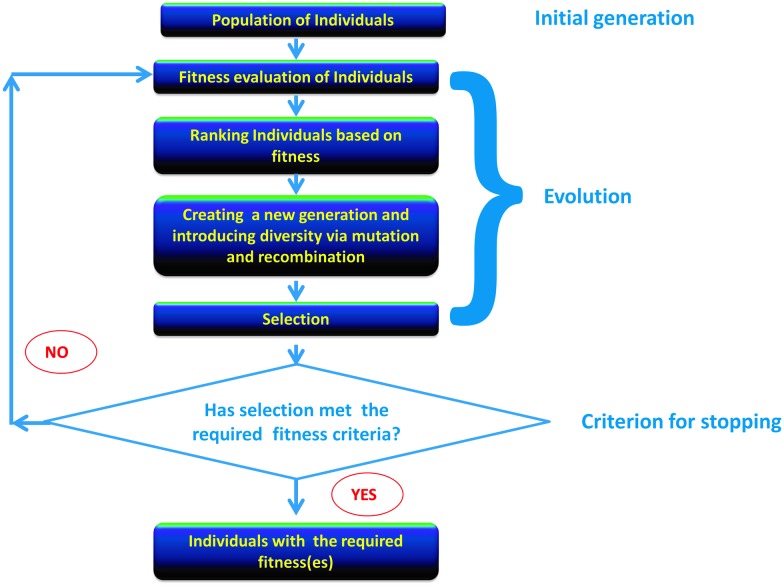
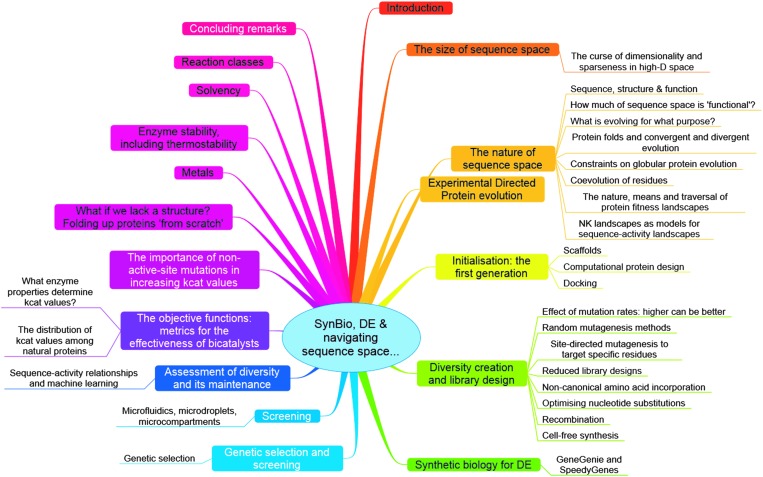
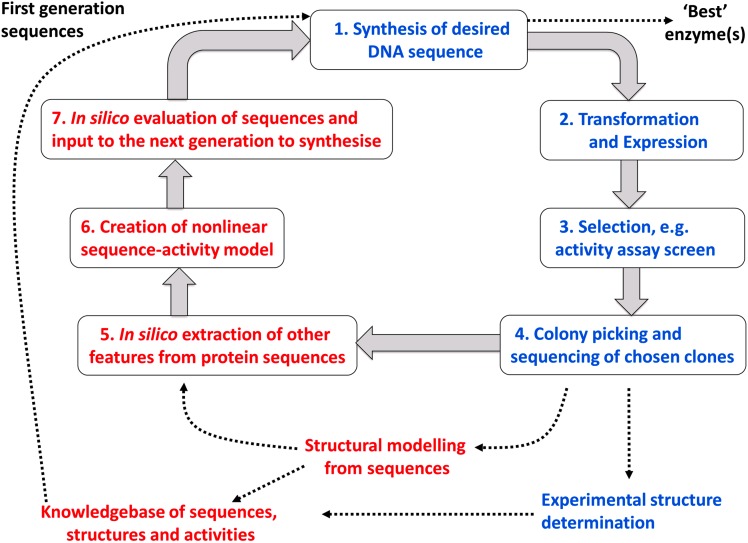
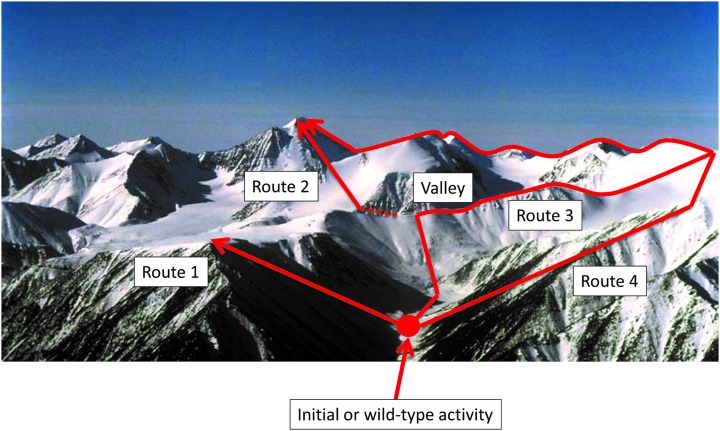
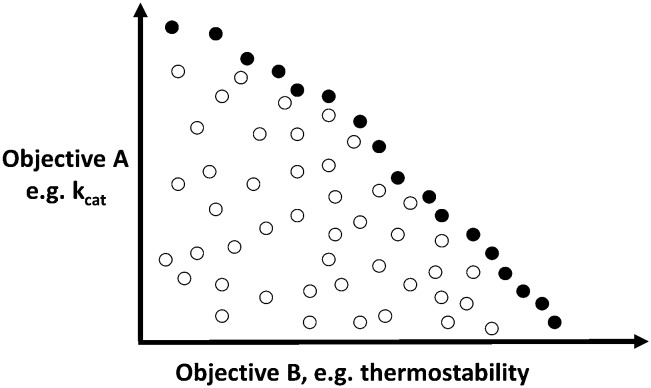
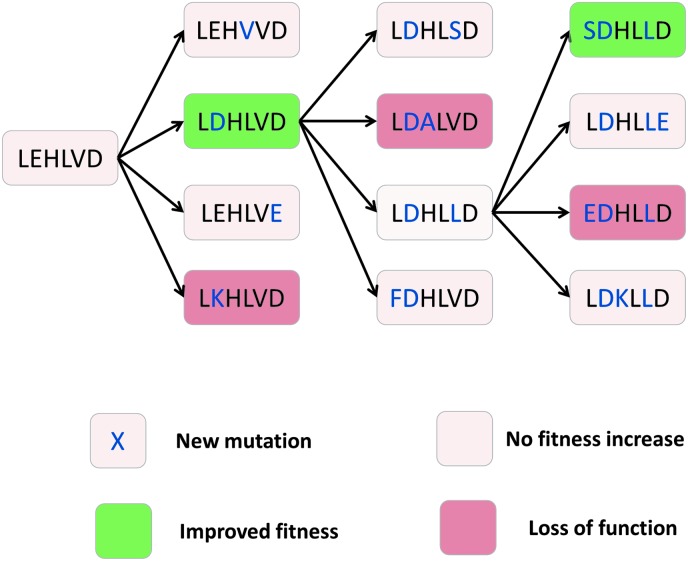
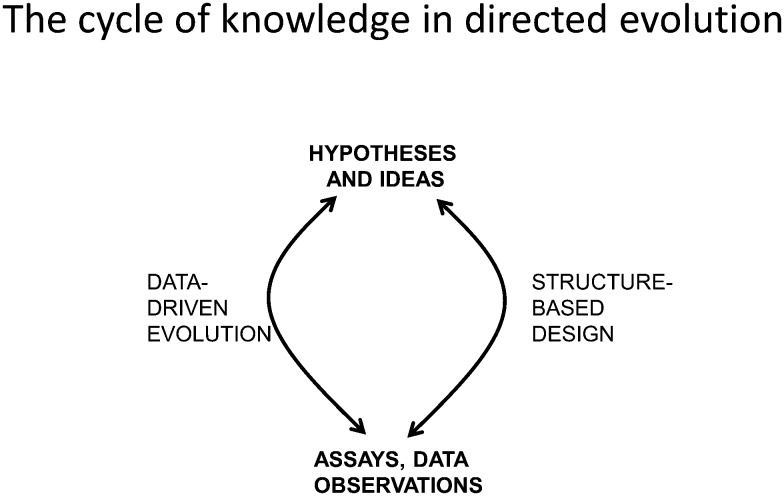
The amino acid sequence of a protein affects both its structure and its function. Thus, the ability to modify the sequence, and hence the structure and activity, of individual proteins in a systematic way, opens up many opportunities, both scientifically and (as we focus on here) for exploitation in biocatalysis. Modern methods of synthetic biology, whereby increasingly large sequences of DNA can be synthesised de novo, allow an unprecedented ability to engineer proteins with novel functions. However, the number of possible proteins is far too large to test individually, so we need means for navigating the 'search space' of possible protein sequences efficiently and reliably in order to find desirable activities and other properties. Enzymologists distinguish binding (Kd) and catalytic (kcat) steps. In a similar way, judicious strategies have blended design (for binding, specificity and active site modelling) with the more empirical methods of classical directed evolution (DE) for improving kcat (where natural evolution rarely seeks the highest values), especially with regard to residues distant from the active site and where the functional linkages underpinning enzyme dynamics are both unknown and hard to predict. Epistasis (where the 'best' amino acid at one site depends on that or those at others) is a notable feature of directed evolution. The aim of this review is to highlight some of the approaches that are being developed to allow us to use directed evolution to improve enzyme properties, often dramatically. We note that directed evolution differs in a number of ways from natural evolution, including in particular the available mechanisms and the likely selection pressures. Thus, we stress the opportunities afforded by techniques that enable one to map sequence to (structure and) activity in silico, as an effective means of modelling and exploring protein landscapes. Because known landscapes may be assessed and reasoned about as a whole, simultaneously, this offers opportunities for protein improvement not readily available to natural evolution on rapid timescales. Intelligent landscape navigation, informed by sequence-activity relationships and coupled to the emerging methods of synthetic biology, offers scope for the development of novel biocatalysts that are both highly active and robust.
Figures
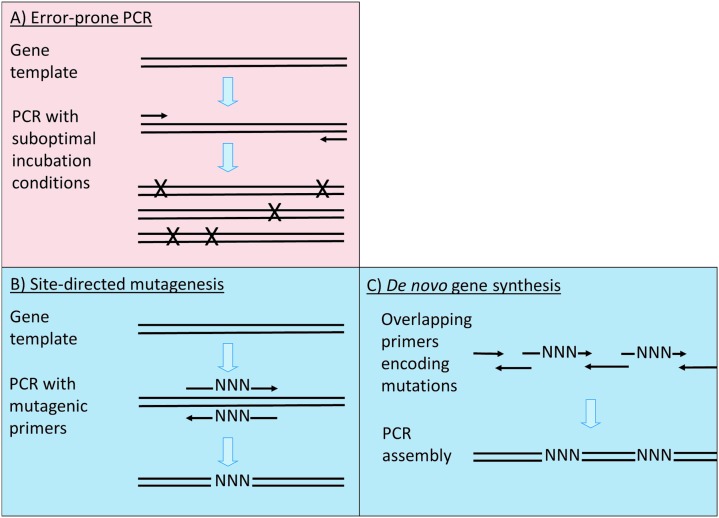
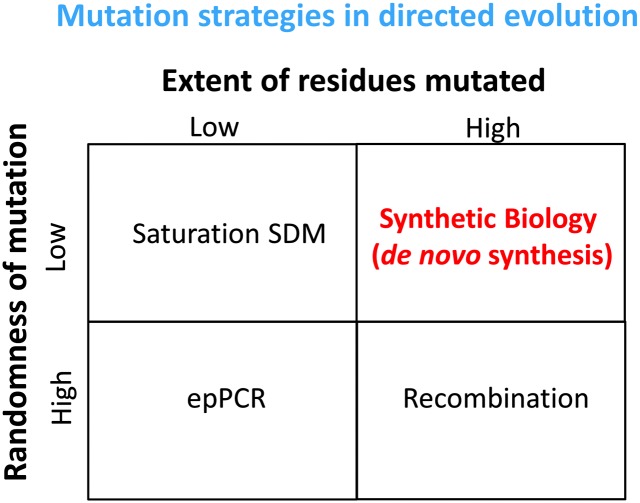
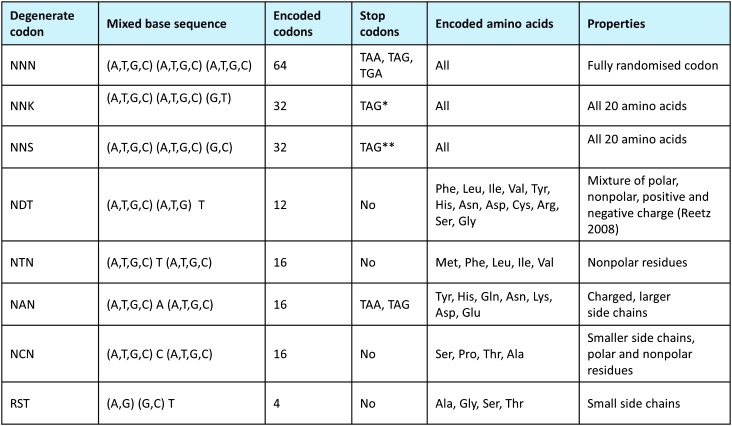
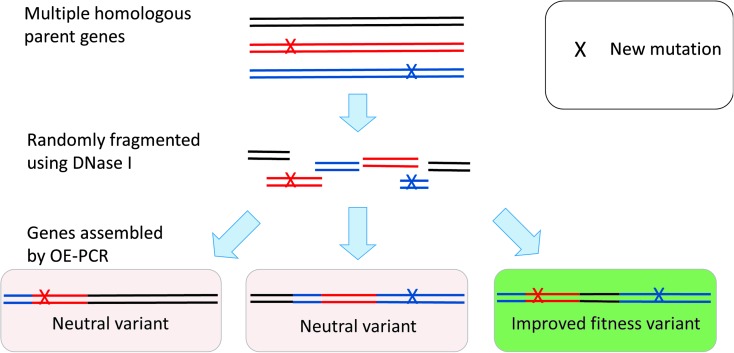
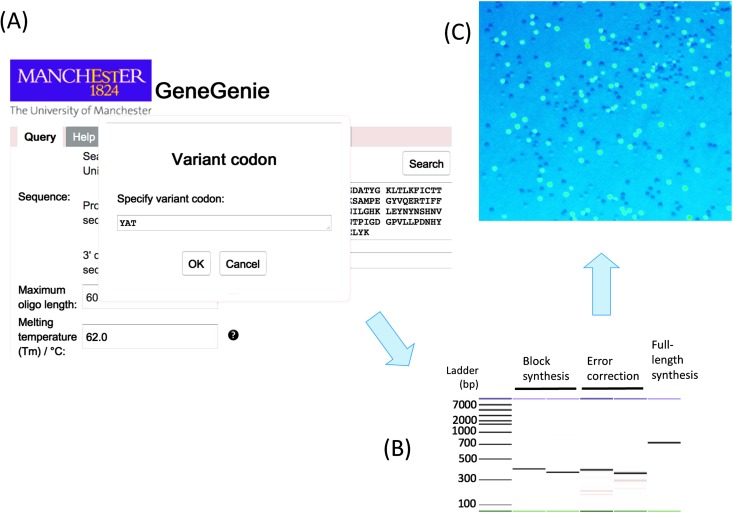
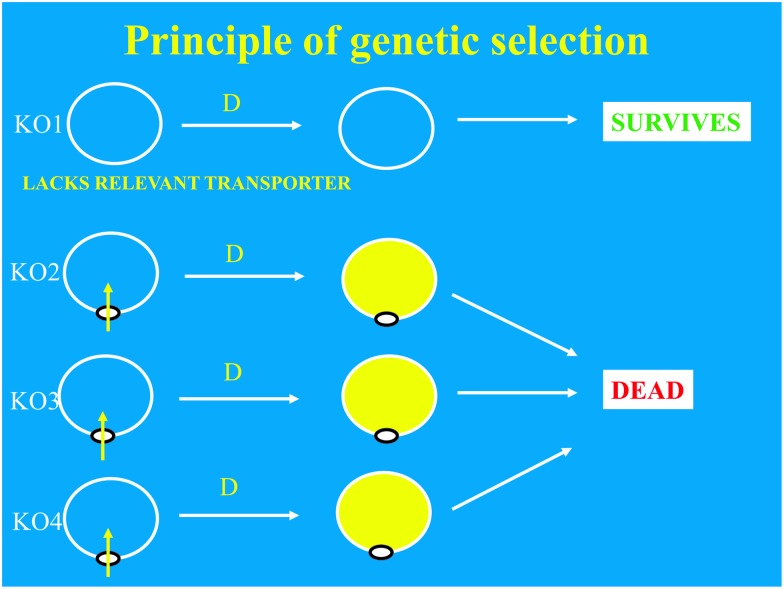
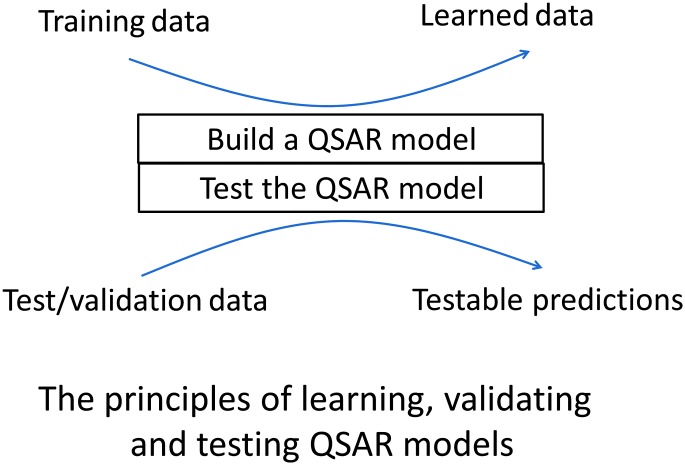
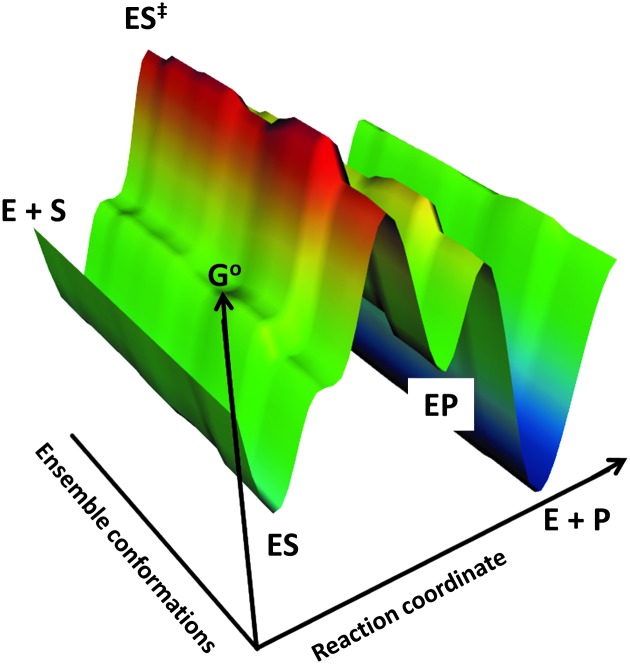




References
-
- Phylogenetic analysis of DNA sequences, ed. M. M. Miyamoto and J. Cracraft, Oxford University Press, Oxford, 1991.
-
- Page R. D. M. and Holmes E. C., Molecular evolution: a phylogenetic approach, Blackwell Science, Oxford, 1998.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources