

High-resolution linkage map and chromosome-scale genome assembly for cassava (Manihot esculenta Crantz) from 10 populations
- PMID: 25504737
- PMCID: PMC4291464
- DOI: 10.1534/g3.114.015008
High-resolution linkage map and chromosome-scale genome assembly for cassava (Manihot esculenta Crantz) from 10 populations
Abstract
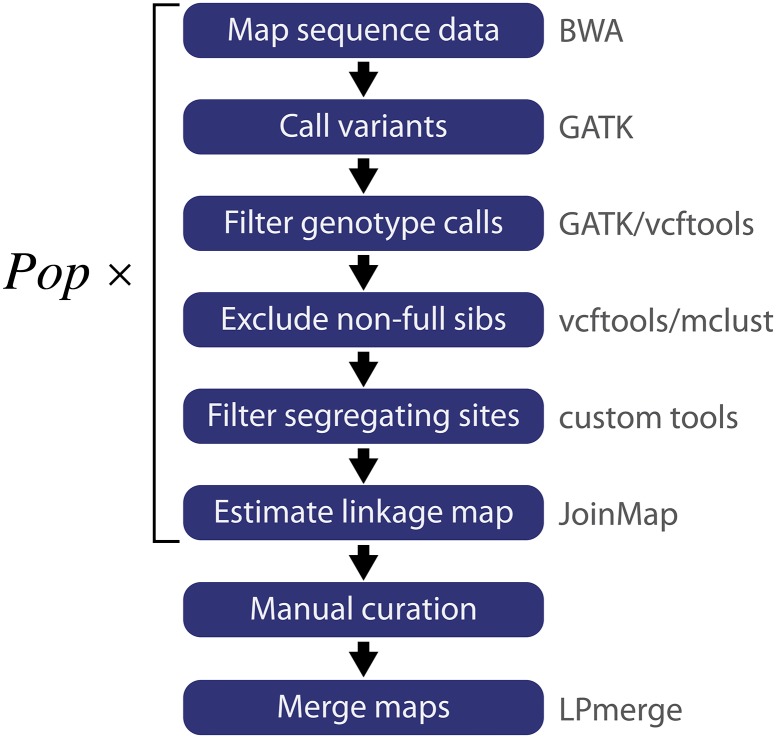
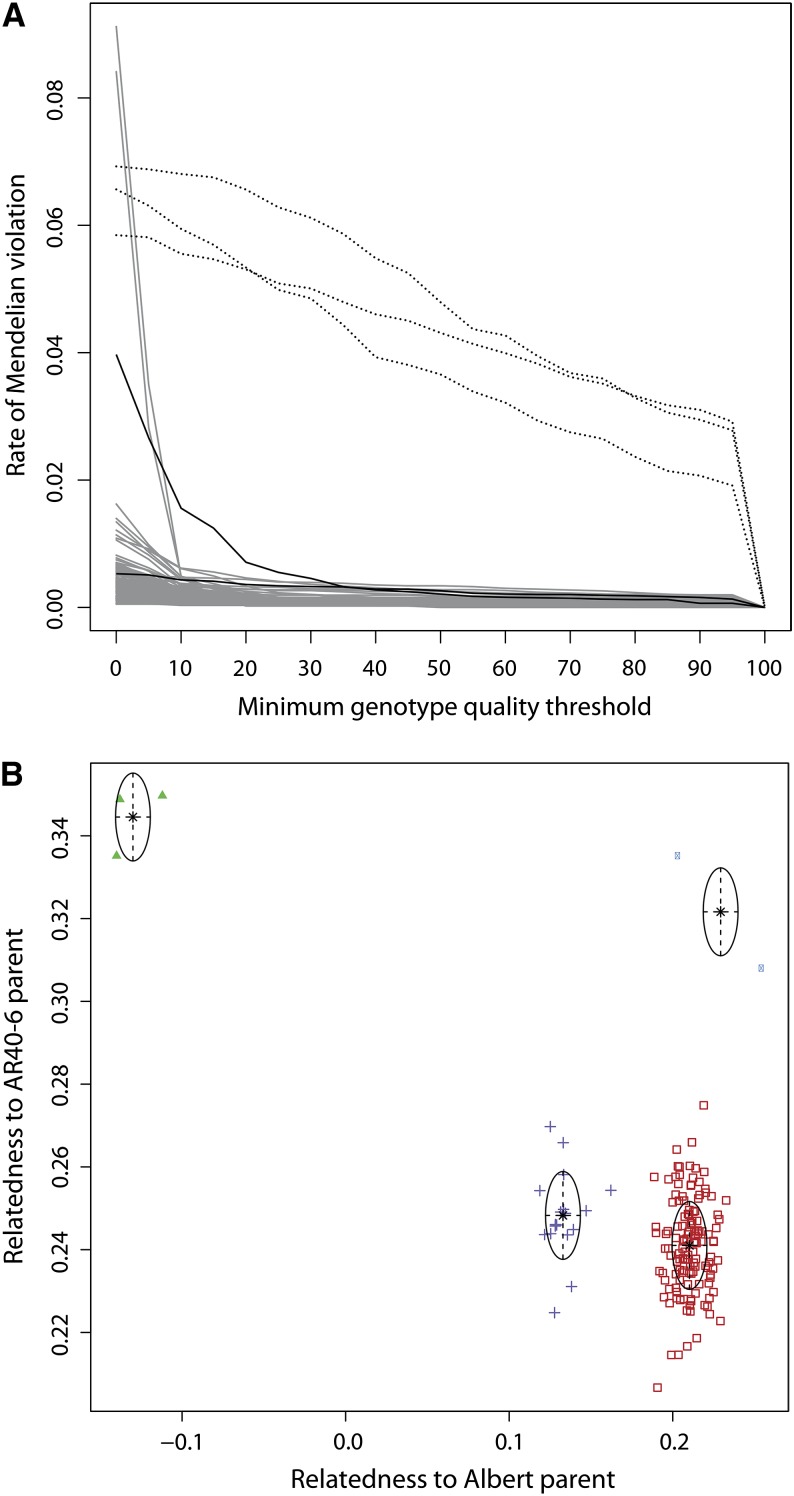
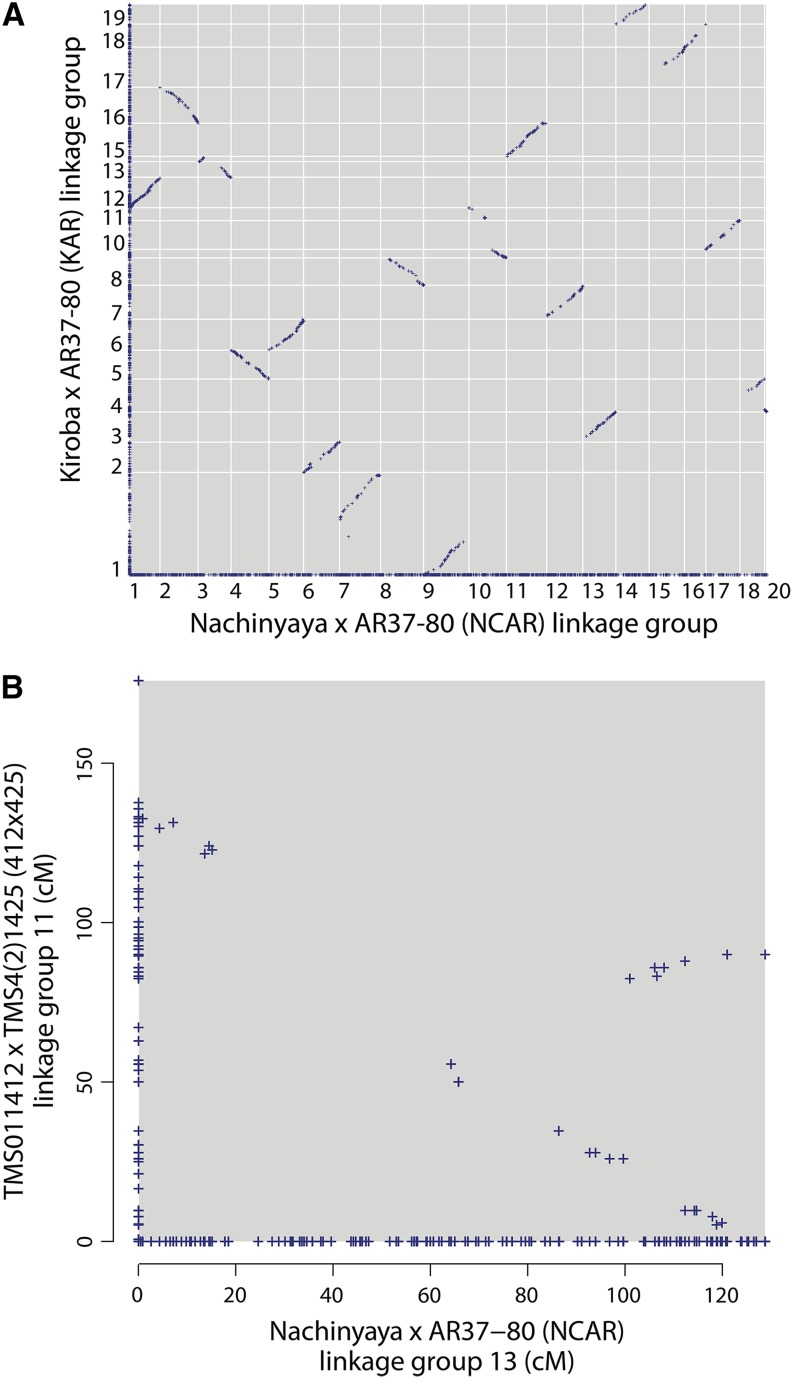
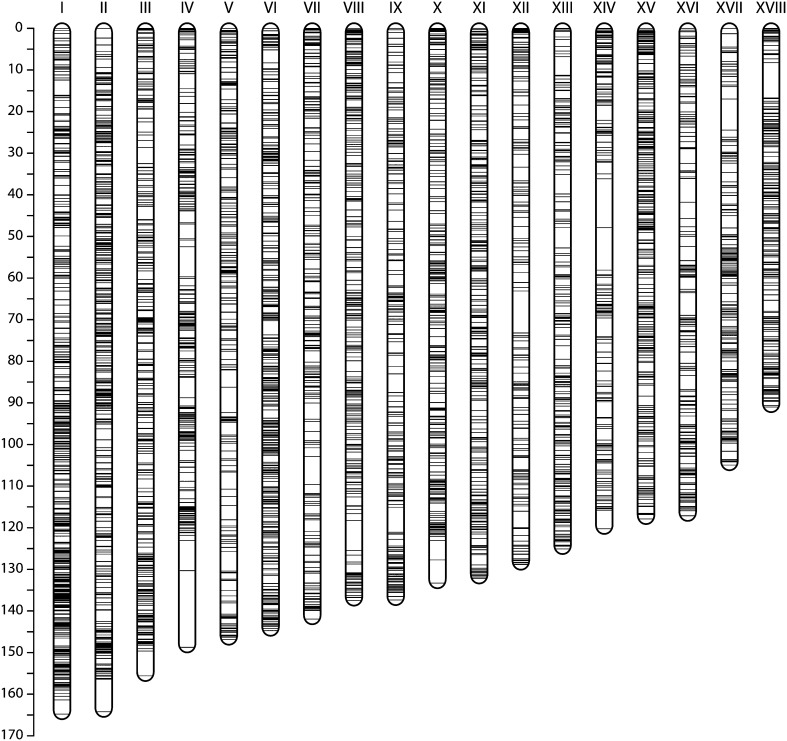
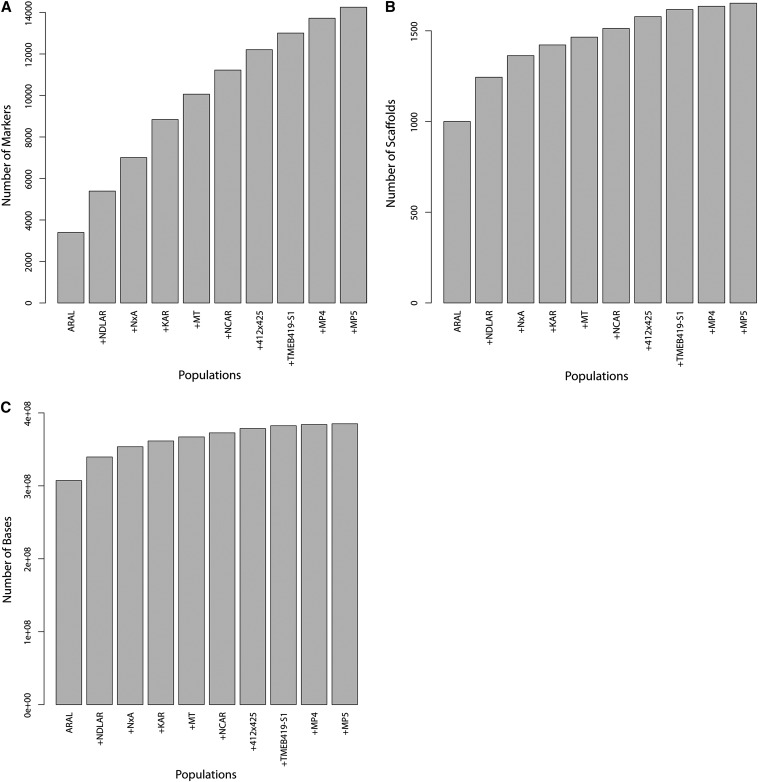
Cassava (Manihot esculenta Crantz) is a major staple crop in Africa, Asia, and South America, and its starchy roots provide nourishment for 800 million people worldwide. Although native to South America, cassava was brought to Africa 400-500 years ago and is now widely cultivated across sub-Saharan Africa, but it is subject to biotic and abiotic stresses. To assist in the rapid identification of markers for pathogen resistance and crop traits, and to accelerate breeding programs, we generated a framework map for M. esculenta Crantz from reduced representation sequencing [genotyping-by-sequencing (GBS)]. The composite 2412-cM map integrates 10 biparental maps (comprising 3480 meioses) and organizes 22,403 genetic markers on 18 chromosomes, in agreement with the observed karyotype. We used the map to anchor 71.9% of the draft genome assembly and 90.7% of the predicted protein-coding genes. The chromosome-anchored genome sequence will be useful for breeding improvement by assisting in the rapid identification of markers linked to important traits, and in providing a framework for genomic selection-enhanced breeding of this important crop.
Keywords: F1 cross; SNP; composite genetic map; genotyping-by-sequencing; pseudomolecules.
Copyright © 2015 International Cassava Genetic Map Consortium (ICGMC).
Figures
References
-
- Altshuler D., Pollara V. J., Cowles C. R., Van Etten W. J., Baldwin J., et al. , 2000. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407: 513–516. - PubMed
-
- Aronesty, E., 2011 Command-line tools for processing biological sequencing data. http://code.google.com/p/ea-utils.
-
- Awoleye F., Duren M., Dolezel J., Novak F. J., 1994. Nuclear DNA content and in vitro induced somatic polyploidization cassava (Manihot esculenta Crantz) breeding. Euphytica 76: 195–202.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources