

mzDB: a file format using multiple indexing strategies for the efficient analysis of large LC-MS/MS and SWATH-MS data sets
- PMID: 25505153
- PMCID: PMC4349994
- DOI: 10.1074/mcp.O114.039115
mzDB: a file format using multiple indexing strategies for the efficient analysis of large LC-MS/MS and SWATH-MS data sets
Abstract
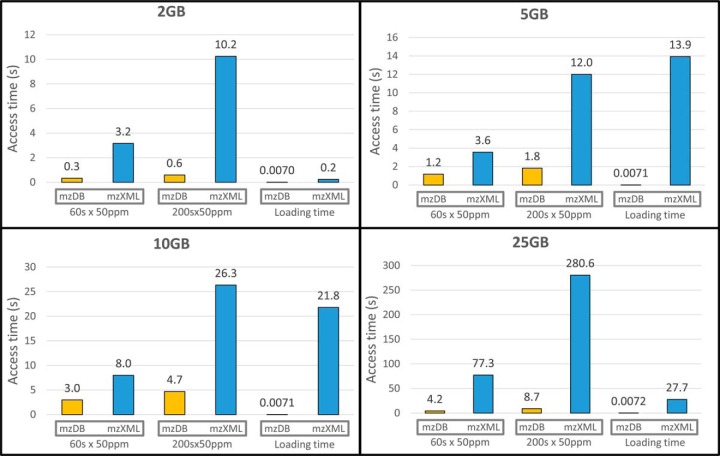
The analysis and management of MS data, especially those generated by data independent MS acquisition, exemplified by SWATH-MS, pose significant challenges for proteomics bioinformatics. The large size and vast amount of information inherent to these data sets need to be properly structured to enable an efficient and straightforward extraction of the signals used to identify specific target peptides. Standard XML based formats are not well suited to large MS data files, for example, those generated by SWATH-MS, and compromise high-throughput data processing and storing. We developed mzDB, an efficient file format for large MS data sets. It relies on the SQLite software library and consists of a standardized and portable server-less single-file database. An optimized 3D indexing approach is adopted, where the LC-MS coordinates (retention time and m/z), along with the precursor m/z for SWATH-MS data, are used to query the database for data extraction. In comparison with XML formats, mzDB saves ∼25% of storage space and improves access times by a factor of twofold up to even 2000-fold, depending on the particular data access. Similarly, mzDB shows also slightly to significantly lower access times in comparison with other formats like mz5. Both C++ and Java implementations, converting raw or XML formats to mzDB and providing access methods, will be released under permissive license. mzDB can be easily accessed by the SQLite C library and its drivers for all major languages, and browsed with existing dedicated GUIs. The mzDB described here can boost existing mass spectrometry data analysis pipelines, offering unprecedented performance in terms of efficiency, portability, compactness, and flexibility.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
Conflict of interest statement: The authors declare no conflict of interest.
Figures
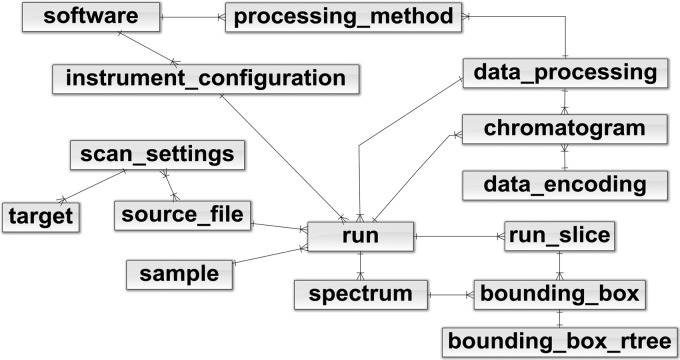
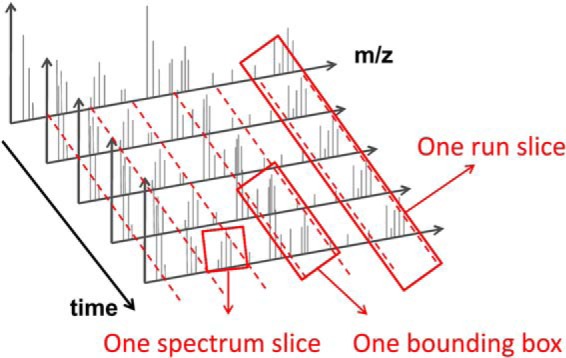
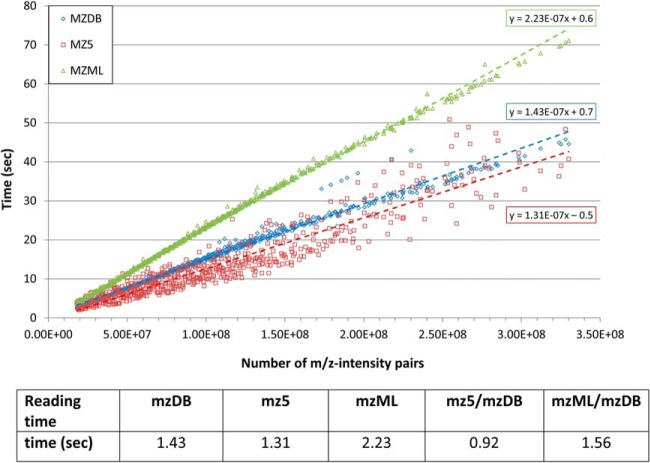
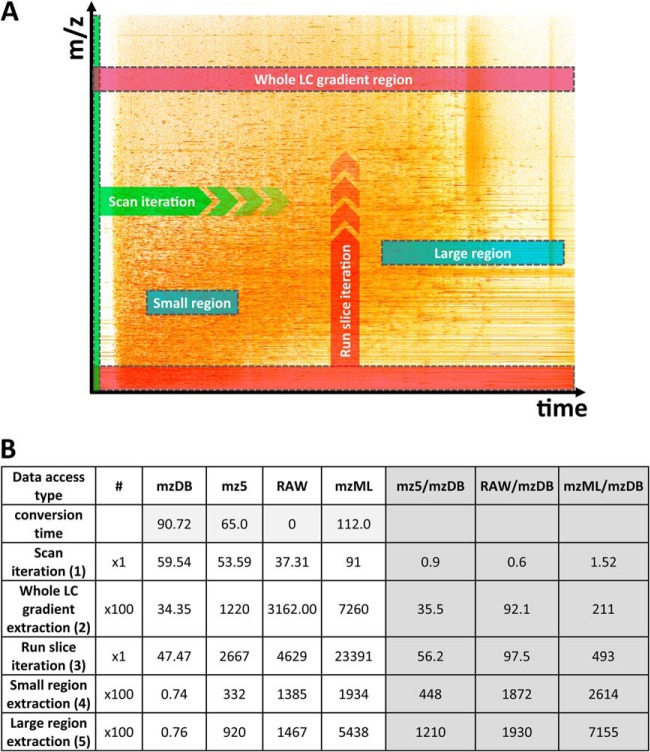
References
-
- Köcher T., Swart R., Mechtler K. (2011) Ultra-high-pressure RPLC hyphenated to an LTQ-Orbitrap Velos reveals a linear relation between peak capacity and number of identified peptides. Anal. Chem. 83, 2699–2704 - PubMed
-
- Nagaraj N., Alexander Kulak N., Cox J., Neuhauser N., Mayr K., Hoerning O., Vorm O., Mann M. (2012) System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteomics 11, M111.013722–M111.013722 - PMC - PubMed
-
- Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
