

IM-TORNADO: a tool for comparison of 16S reads from paired-end libraries
- PMID: 25506826
- PMCID: PMC4266640
- DOI: 10.1371/journal.pone.0114804
IM-TORNADO: a tool for comparison of 16S reads from paired-end libraries
Abstract
Motivation: 16S rDNA hypervariable tag sequencing has become the de facto method for accessing microbial diversity. Illumina paired-end sequencing, which produces two separate reads for each DNA fragment, has become the platform of choice for this application. However, when the two reads do not overlap, existing computational pipelines analyze data from read separately and underutilize the information contained in the paired-end reads.
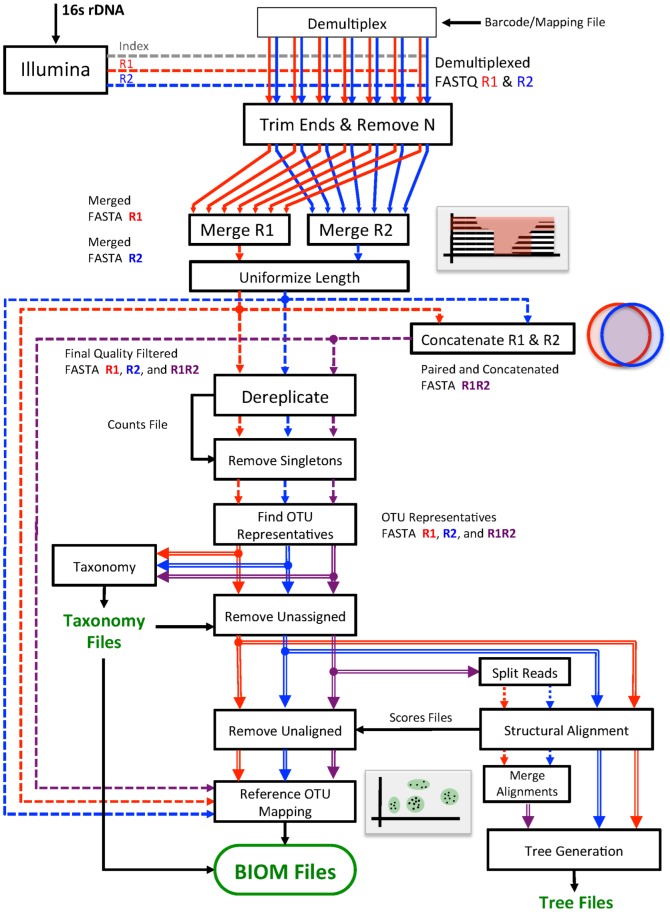
Results: We created a workflow known as Illinois Mayo Taxon Organization from RNA Dataset Operations (IM-TORNADO) for processing non-overlapping reads while retaining maximal information content. Using synthetic mock datasets, we show that the use of both reads produced answers with greater correlation to those from full length 16S rDNA when looking at taxonomy, phylogeny, and beta-diversity.
Availability and implementation: IM-TORNADO is freely available at http://sourceforge.net/projects/imtornado and produces BIOM format output for cross compatibility with other pipelines such as QIIME, mothur, and phyloseq.
Conflict of interest statement
Figures
References
-
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
