

Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer's Disease: Understanding the Therapeutics Strategies
- PMID: 25511446
- PMCID: PMC4470891
- DOI: 10.1007/s12035-014-9053-6
Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer's Disease: Understanding the Therapeutics Strategies
Abstract
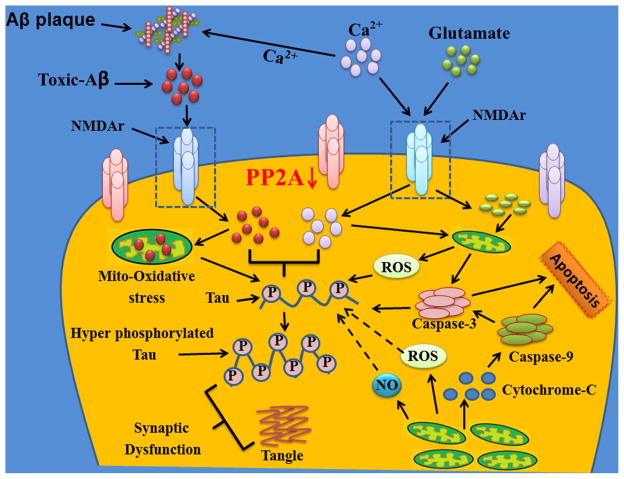
Synapses are formed by interneuronal connections that permit a neuronal cell to pass an electrical or chemical signal to another cell. This passage usually gets damaged or lost in most of the neurodegenerative diseases. It is widely believed that the synaptic dysfunction and synapse loss contribute to the cognitive deficits in patients with Alzheimer's disease (AD). Although pathological hallmarks of AD are senile plaques, neurofibrillary tangles, and neuronal degeneration which are associated with increased oxidative stress, synaptic loss is an early event in the pathogenesis of AD. The involvement of major kinases such as mitogen-activated protein kinase (MAPK), extracellular receptor kinase (ERK), calmodulin-dependent protein kinase (CaMKII), glycogen synthase-3β (GSK-3β), cAMP response element-binding protein (CREB), and calcineurin is dynamically associated with oxidative stress-mediated abnormal hyperphosphorylation of tau and suggests that alteration of these kinases could exclusively be involved in the pathogenesis of AD. N-methyl-D-aspartate (NMDA) receptor (NMDAR) activation and beta amyloid (Aβ) toxicity alter the synapse function, which is also associated with protein phosphatase (PP) inhibition and tau hyperphosphorylation (two main events of AD). However, the involvement of oxidative stress in synapse dysfunction is poorly understood. Oxidative stress and free radical generation in the brain along with excitotoxicity leads to neuronal cell death. It is inferred from several studies that excitotoxicity, free radical generation, and altered synaptic function encouraged by oxidative stress are associated with AD pathology. NMDARs maintain neuronal excitability, Ca(2+) influx, and memory formation through mechanisms of synaptic plasticity. Recently, we have reported the mechanism of the synapse redox stress associated with NMDARs altered expression. We suggest that oxidative stress mediated through NMDAR and their interaction with other molecules might be a driving force for tau hyperphosphorylation and synapse dysfunction. Thus, understanding the oxidative stress mechanism and degenerating synapses is crucial for the development of therapeutic strategies designed to prevent AD pathogenesis.
Keywords: Alzheimer’s disease; Kinases; NMDA receptor; Oxidative stress; Synaptic function; Tau protein.
Conflict of interest statement
Figures




References
-
- Wang Z, Yang L, Zheng H. Role of APP and Abeta in synaptic physiology. Curr Alzheimer Res. 2012;9:217–226. - PubMed
-
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. - PubMed
-
- Rai S, Kamat PK, Nath C, Shukla R. Glial activation and post-synaptic neurotoxicity: the key events in streptozotocin (ICV) induced memory impairment in rats. Pharmacol Biochem Behav. 2014;117:104–117. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
