

Prehistoric genomes reveal the genetic foundation and cost of horse domestication
- PMID: 25512547
- PMCID: PMC4284583
- DOI: 10.1073/pnas.1416991111
Prehistoric genomes reveal the genetic foundation and cost of horse domestication
Abstract
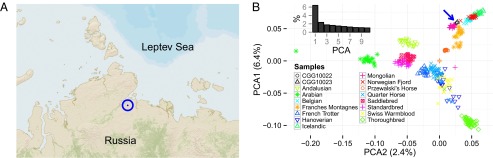
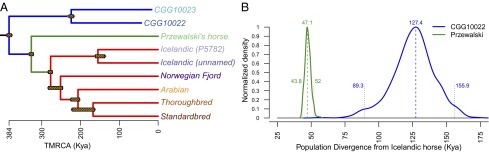
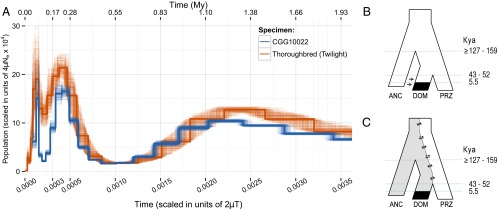
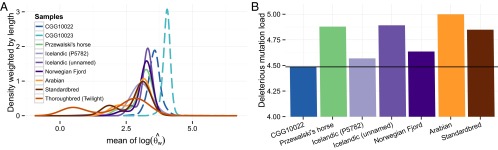
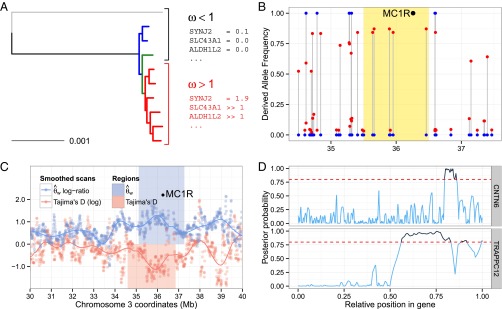
The domestication of the horse ∼ 5.5 kya and the emergence of mounted riding, chariotry, and cavalry dramatically transformed human civilization. However, the genetics underlying horse domestication are difficult to reconstruct, given the near extinction of wild horses. We therefore sequenced two ancient horse genomes from Taymyr, Russia (at 7.4- and 24.3-fold coverage), both predating the earliest archeological evidence of domestication. We compared these genomes with genomes of domesticated horses and the wild Przewalski's horse and found genetic structure within Eurasia in the Late Pleistocene, with the ancient population contributing significantly to the genetic variation of domesticated breeds. We furthermore identified a conservative set of 125 potential domestication targets using four complementary scans for genes that have undergone positive selection. One group of genes is involved in muscular and limb development, articular junctions, and the cardiac system, and may represent physiological adaptations to human utilization. A second group consists of genes with cognitive functions, including social behavior, learning capabilities, fear response, and agreeableness, which may have been key for taming horses. We also found that domestication is associated with inbreeding and an excess of deleterious mutations. This genetic load is in line with the "cost of domestication" hypothesis also reported for rice, tomatoes, and dogs, and it is generally attributed to the relaxation of purifying selection resulting from the strong demographic bottlenecks accompanying domestication. Our work demonstrates the power of ancient genomes to reconstruct the complex genetic changes that transformed wild animals into their domesticated forms, and the population context in which this process took place.
Keywords: Przewalski’s horse; ancient DNA; cost of domestication; horse domestication; positive selection.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Kelekna P. The Horse in Human History. Cambridge Univ Press; Cambridge, England: 2009.
-
- Outram AK, et al. The earliest horse harnessing and milking. Science. 2009;323(5919):1332–1335. - PubMed
-
- Cruz F, Vilà C, Webster MT. The legacy of domestication: Accumulation of deleterious mutations in the dog genome. Mol Biol Evol. 2008;25(11):2331–2336. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
