

The optimal corepressor function of nuclear receptor corepressor (NCoR) for peroxisome proliferator-activated receptor γ requires G protein pathway suppressor 2
- PMID: 25519902
- PMCID: PMC4319032
- DOI: 10.1074/jbc.M114.598797
The optimal corepressor function of nuclear receptor corepressor (NCoR) for peroxisome proliferator-activated receptor γ requires G protein pathway suppressor 2
Abstract
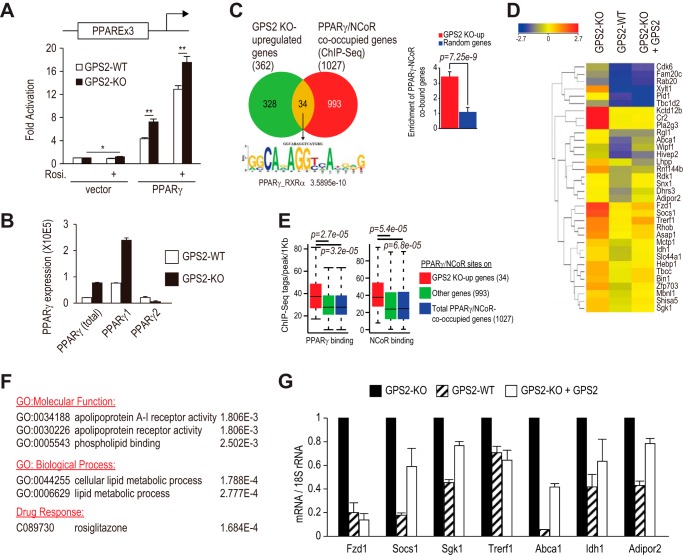
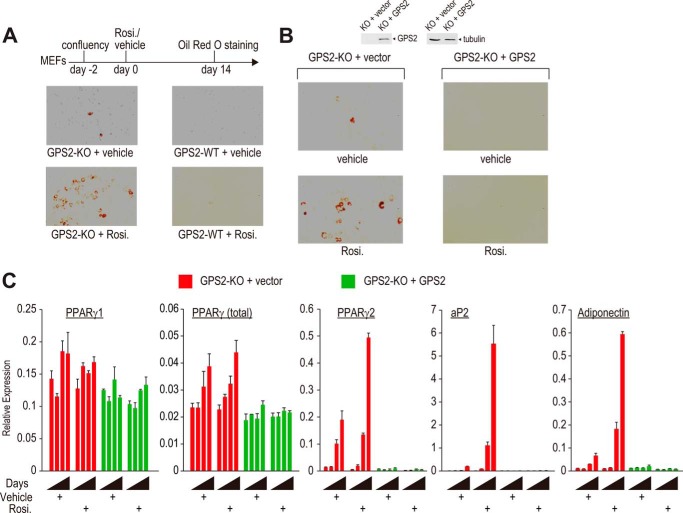
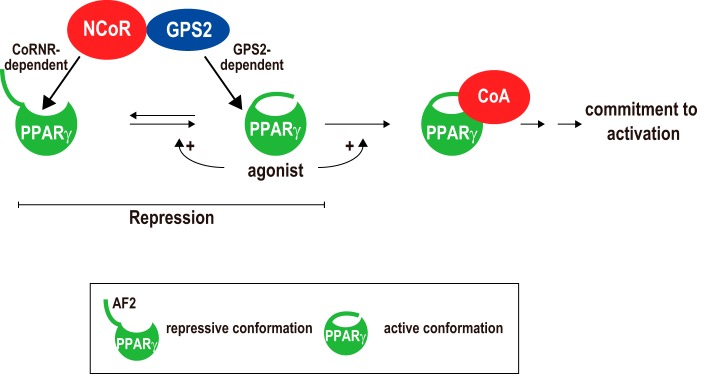
Repression of peroxisome proliferator-activated receptor γ (PPARγ)-dependent transcription by the nuclear receptor corepressor (NCoR) is important for homeostatic expression of PPARγ target genes in vivo. The current model states that NCoR-mediated repression requires its direct interaction with PPARγ in the repressive conformation. Previous studies, however, have shown that DNA-bound PPARγ is incompatible with a direct, high-affinity association with NCoR because of the inherent ability of PPARγ to adopt the active conformation. Here we show that NCoR acquires the ability to repress active PPARγ-mediated transcription via G protein pathway suppressor 2 (GPS2), a component of the NCoR corepressor complex. Unlike NCoR, GPS2 can recognize and bind the active state of PPARγ. In GPS2-deficient mouse embryonic fibroblast cells, loss of GPS2 markedly reduces the corepressor function of NCoR for PPARγ, leading to constitutive activation of PPARγ target genes and spontaneous adipogenesis of the cells. GPS2, however, is dispensable for repression mediated by unliganded thyroid hormone receptor α or a PPARγ mutant unable to adopt the active conformation. This study shows that GPS2, although dispensable for the intrinsic repression function of NCoR, can mediate a novel corepressor repression pathway that allows NCoR to directly repress active PPARγ-mediated transcription, which is important for the optimal corepressor function of NCoR for PPARγ. Interestingly, GPS2-dependent repression specifically targets PPARγ but not PPARα or PPARδ. Therefore, GPS2 may serve as a unique target to manipulate PPARγ signaling in diseases.
Keywords: Conformational Change; GPS2; NCoR; Nuclear Receptor; PPARγ; Peroxisome Proliferator-Activated Receptor (PPAR); Thyroid Hormone; Transcription Coactivator; Transcription Corepressor; Transcriptional Repression.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Glass C. K., Rosenfeld M. G. (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14, 121–141 - PubMed
-
- Bourguet W., Ruff M., Chambon P., Gronemeyer H., Moras D. (1995) Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-α. Nature 375, 377–382 - PubMed
-
- Wagner R. L., Apriletti J. W., McGrath M. E., West B. L., Baxter J. D., Fletterick R. J. (1995) A structural role for hormone in the thyroid hormone receptor. Nature 378, 690–697 - PubMed
-
- Perissi V., Rosenfeld M. G. (2005) Controlling nuclear receptors: the circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 6, 542–554 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
