

Successful control with carbamazepine of family with paroxysmal kinesigenic dyskinesia of PRRT2 mutation
- PMID: 25520928
- PMCID: PMC4265013
- DOI: 10.7603/s40681-014-0015-0
Successful control with carbamazepine of family with paroxysmal kinesigenic dyskinesia of PRRT2 mutation
Abstract
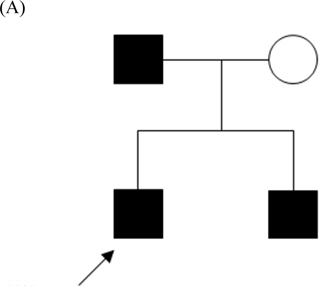
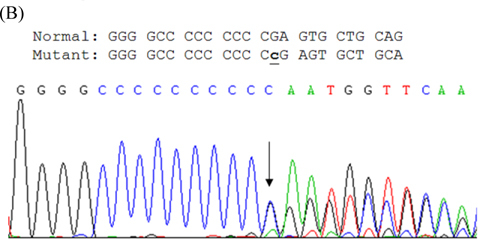
Paroxysmal kinesigenic dyskinesia (PKD), a rare paroxysmal movement disorder often misdiagnosed as epilepsy, is characterized by recurrent, brief dyskinesia attacks triggered by sudden voluntary movement. Pathophysiological mechanism of PKD remains not well understood. Ion channelopathy has been suggested, since the disease responds well to ion channel blockers. Mutations in proline-rich transmembrane protein 2 (PRRT2) were recently identified in patients with familial PKD. To extend these genetic reports, we studied a family with clinical manifestations of familial PKD responding well to low dose carbamazepine. Therapeutic dose ranged from 1.5 to 2.0 mg/ kg/day, below that in seizure control. One insertion mutation c.649_650insC (p.P217fsX7) was identified in three patients of the family. This study avers PRRT2's high sensitivity for PKD phenotype. Identification of genes underlying pathogenesis will enhance diagnosis and treatment. Function of PRRT2 and its role in PKD warrant further investigation.
Keywords: Carbamazepin; Mutation; PRRT2; Paroxysmal dyskinesia.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources