

Altered chromatin occupancy of master regulators underlies evolutionary divergence in the transcriptional landscape of erythroid differentiation
- PMID: 25521328
- PMCID: PMC4270484
- DOI: 10.1371/journal.pgen.1004890
Altered chromatin occupancy of master regulators underlies evolutionary divergence in the transcriptional landscape of erythroid differentiation
Abstract
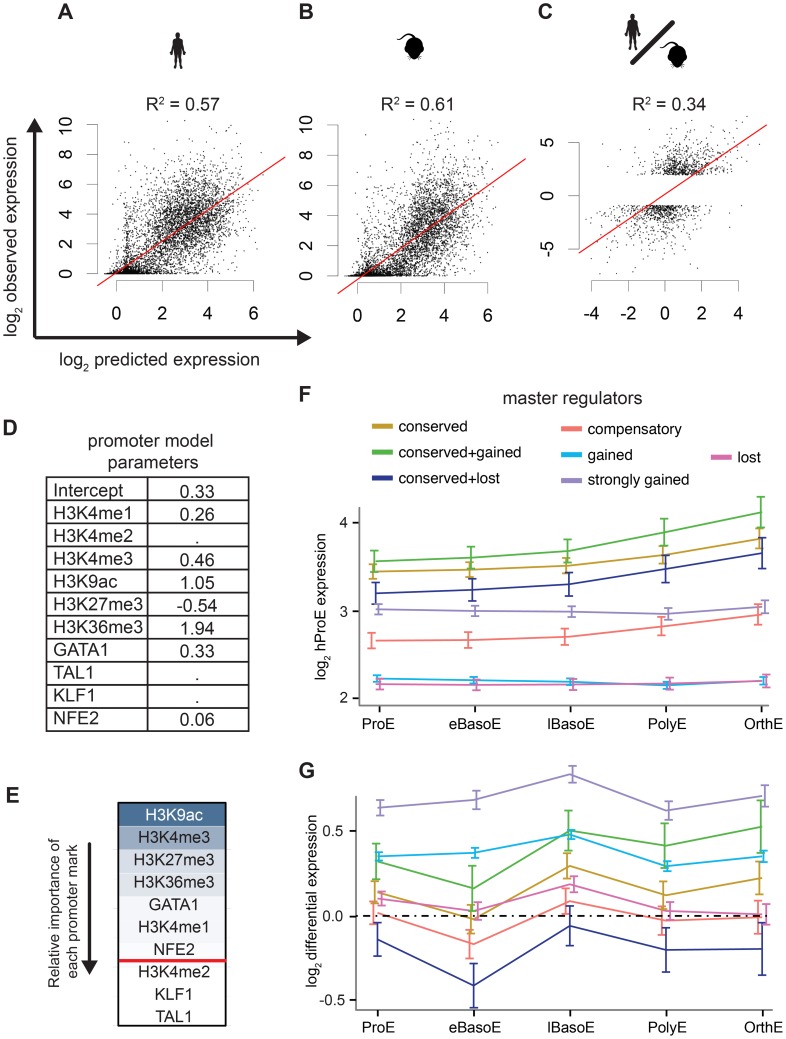
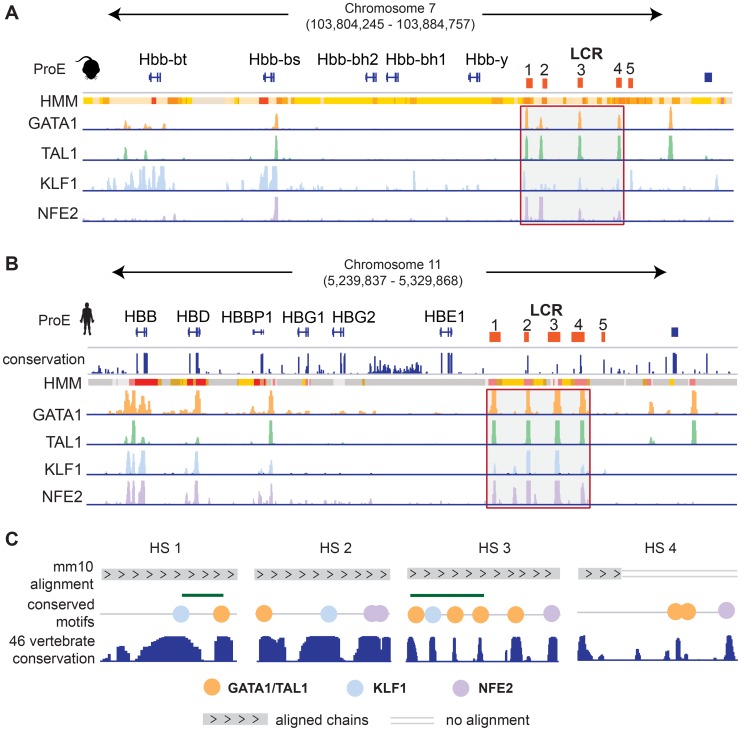
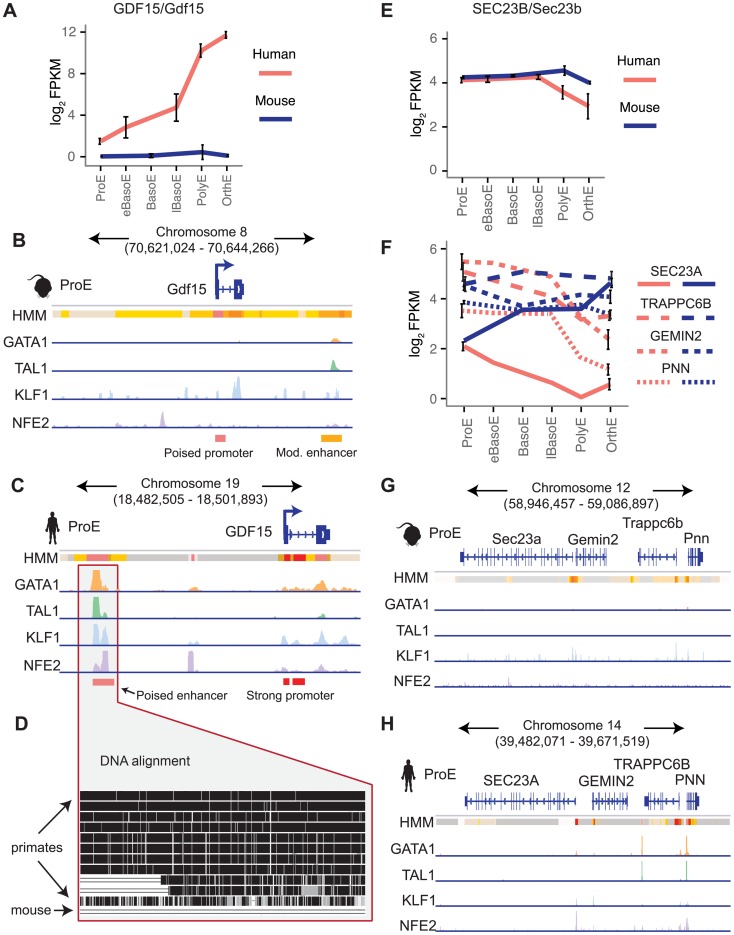
Erythropoiesis is one of the best understood examples of cellular differentiation. Morphologically, erythroid differentiation proceeds in a nearly identical fashion between humans and mice, but recent evidence has shown that networks of gene expression governing this process are divergent between species. We undertook a systematic comparative analysis of six histone modifications and four transcriptional master regulators in primary proerythroblasts and erythroid cell lines to better understand the underlying basis of these transcriptional differences. Our analyses suggest that while chromatin structure across orthologous promoters is strongly conserved, subtle differences are associated with transcriptional divergence between species. Many transcription factor (TF) occupancy sites were poorly conserved across species (∼25% for GATA1, TAL1, and NFE2) but were more conserved between proerythroblasts and cell lines derived from the same species. We found that certain cis-regulatory modules co-occupied by GATA1, TAL1, and KLF1 are under strict evolutionary constraint and localize to genes necessary for erythroid cell identity. More generally, we show that conserved TF occupancy sites are indicative of active regulatory regions and strong gene expression that is sustained during maturation. Our results suggest that evolutionary turnover of TF binding sites associates with changes in the underlying chromatin structure, driving transcriptional divergence. We provide examples of how this framework can be applied to understand epigenomic variation in specific regulatory regions, such as the β-globin gene locus. Our findings have important implications for understanding epigenomic changes that mediate variation in cellular differentiation across species, while also providing a valuable resource for studies of hematopoiesis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
