

Mitochondrial DNA: impacting central and peripheral nervous systems
- PMID: 25521375
- PMCID: PMC4271190
- DOI: 10.1016/j.neuron.2014.11.022
Mitochondrial DNA: impacting central and peripheral nervous systems
Abstract
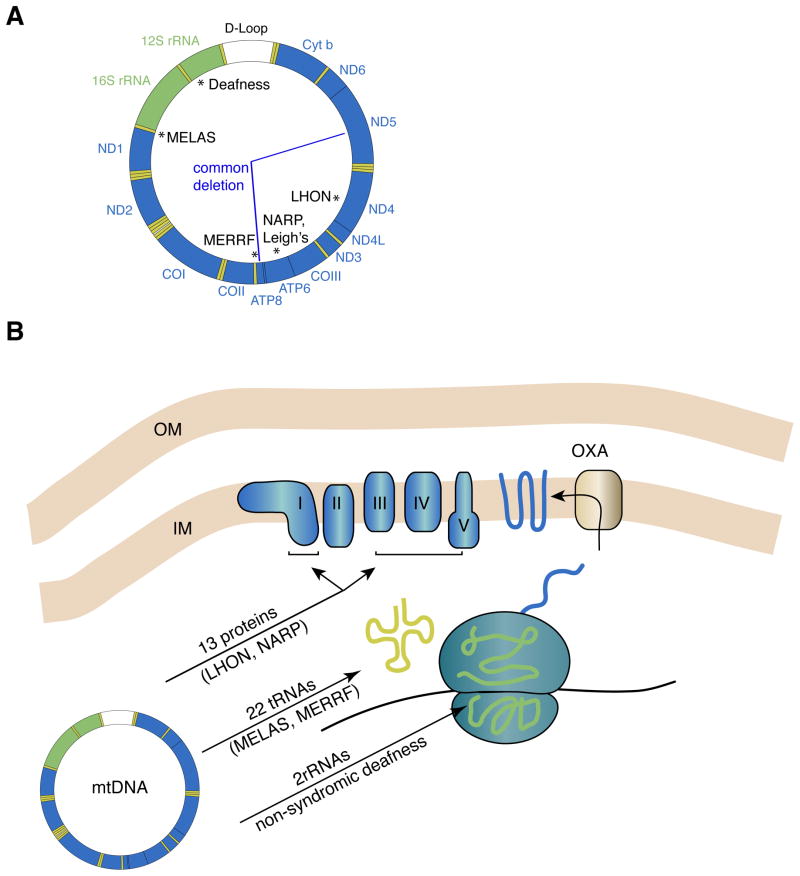
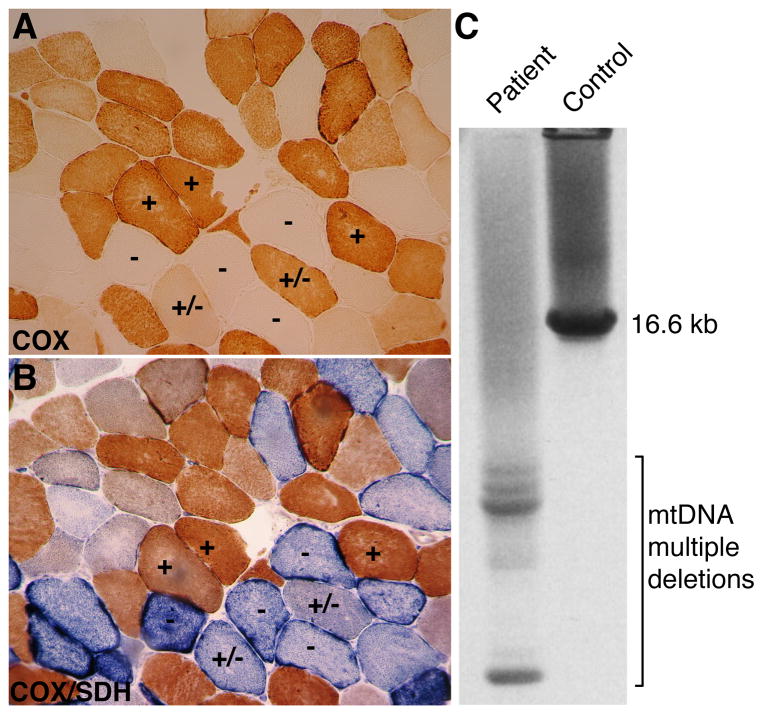
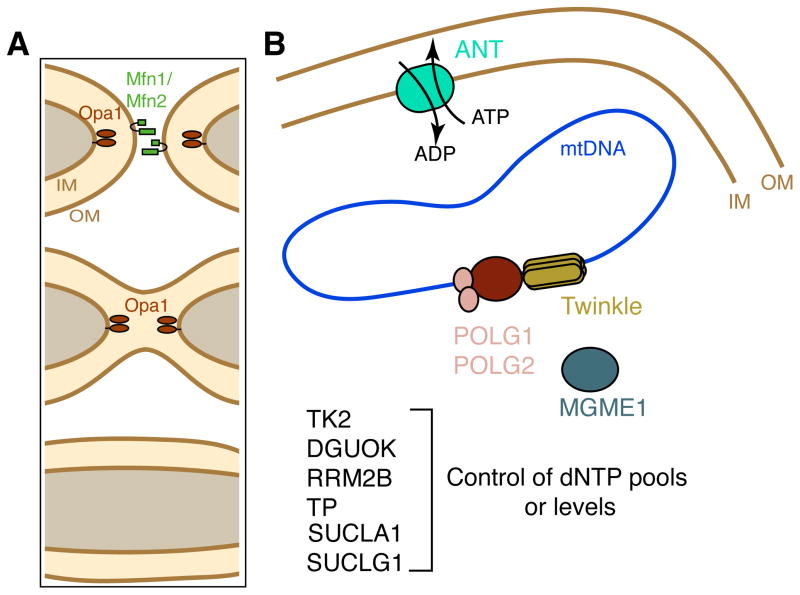
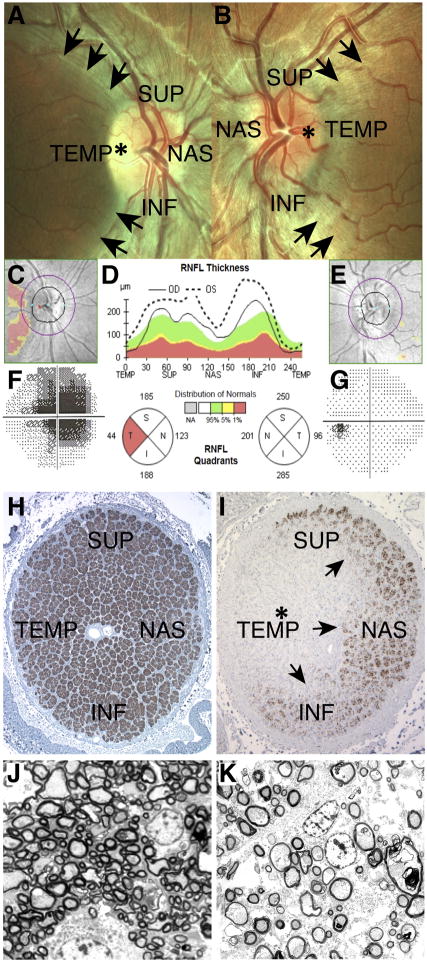
Because of their high-energy metabolism, neurons are strictly dependent on mitochondria, which generate cellular ATP through oxidative phosphorylation. The mitochondrial genome encodes for critical components of the oxidative phosphorylation pathway machinery, and therefore, mutations in mitochondrial DNA (mtDNA) cause energy production defects that frequently have severe neurological manifestations. Here, we review the principles of mitochondrial genetics and focus on prototypical mitochondrial diseases to illustrate how primary defects in mtDNA or secondary defects in mtDNA due to nuclear genome mutations can cause prominent neurological and multisystem features. In addition, we discuss the pathophysiological mechanisms underlying mitochondrial diseases, the cellular mechanisms that protect mitochondrial integrity, and the prospects for therapy.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
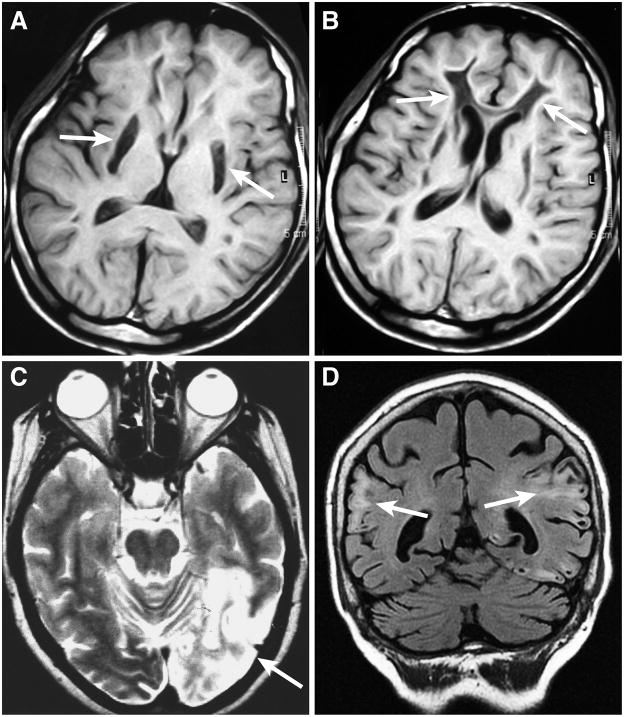
References
-
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nature reviews Neuroscience. 2006;7:207–219. - PubMed
-
- Achilli A, Iommarini L, Olivieri A, Pala M, Hooshiar Kashani B, Reynier P, La Morgia C, Valentino ML, Liguori R, Pizza F, Barboni P, Sadun F, De Negri AM, Zeviani M, Dollfus H, Moulignier A, Ducos G, Orssaud C, Bonneau D, Procaccio V, Leo-Kottler B, Fauser S, Wissinger B, Amati-Bonneau P, Torroni A, Carelli V. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber’s hereditary optic neuropathy. PLoS One. 2012:7. - PMC - PubMed
-
- Ahlqvist KJ, Hamalainen RH, Yatsuga S, Uutela M, Terzioglu M, Gotz A, Forsstrom S, Salven P, Angers-Loustau A, Kopra OH, Tyynismaa H, Larsson NG, Wartiovaara K, Prolla T, Trifunovic A, Suomalainen A. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell metabolism. 2012;15:100–109. - PubMed
-
- Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. - PubMed
-
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, Labauge P, Figarella-Branger D, Marcorelles P, Furby A, Beauvais K, Letournel F, Liguori R, La Morgia C, Montagna P, Liguori M, Zanna C, Rugolo M, Cossarizza A, Wissinger B, Verny C, Schwarzenbacher R, Martin MA, Arenas J, Ayuso C, Garesse R, Lenaers G, Bonneau D, Carelli V. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
