

Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer's disease
- PMID: 25523424
- PMCID: PMC4820400
- DOI: 10.2174/1567205012666141218140953
Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer's disease
Abstract
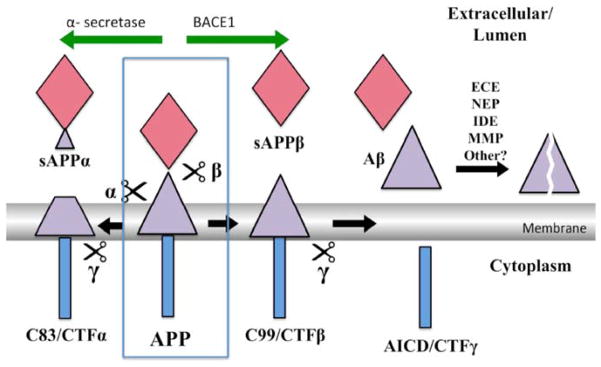
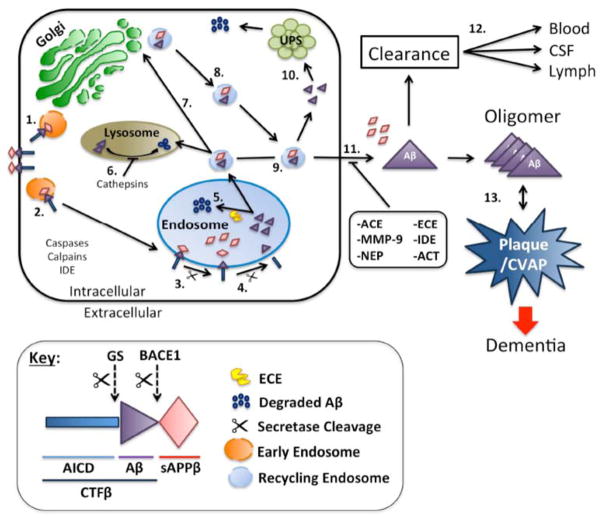
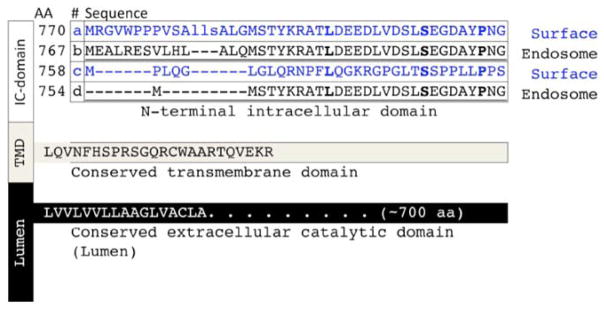
Amyloid-β proteins (Aβ) of 42 (Aβ42) and 40 aa (Aβ40) accumulate as senile plaques (SP) and cerebrovascular amyloid protein deposits that are defining diagnostic features of Alzheimer's disease (AD). A number of rare mutations linked to familial AD (FAD) on the Aβ precursor protein (APP), Presenilin-1 (PS1), Presenilin- 2 (PS2), Adamalysin10, and other genetic risk factors for sporadic AD such as the ε4 allele of Apolipoprotein E (ApoE-ε4) foster the accumulation of Aβ and also induce the entire spectrum of pathology associated with the disease. Aβ accumulation is therefore a key pathological event and a prime target for the prevention and treatment of AD. APP is sequentially processed by β-site APP cleaving enzyme (BACE1) and γ-secretase, a multisubunit PS1/PS2-containing integral membrane protease, to generate Aβ. Although Aβ accumulates in all forms of AD, the only pathways known to be affected in FAD increase Aβ production by APP gene duplication or via base substitutions on APP and γ-secretase subunits PS1 and PS2 that either specifically increase the yield of the longer Aβ42 or both Aβ40 and Aβ42. However, the vast majority of AD patients accumulate Aβ without these known mutations. This led to proposals that impairment of Aβ degradation or clearance may play a key role in AD pathogenesis. Several candidate enzymes, including Insulin-degrading enzyme (IDE), Neprilysin (NEP), Endothelin-converting enzyme (ECE), Angiotensin converting enzyme (ACE), Plasmin, and Matrix metalloproteinases (MMPs) have been identified and some have even been successfully evaluated in animal models. Several studies also have demonstrated the capacity of γ-secretase inhibitors to paradoxically increase the yield of Aβ and we have recently established that the mechanism is by skirting Aβ degradation. This review outlines major cellular pathways of Aβ degradation to provide a basis for future efforts to fully characterize the panel of pathways responsible for Aβ turnover.
Conflict of interest statement
The authors confirm that this article content has no conflict of interest.
Figures
References
-
- Thies W, Bleiler L, Alzheimer’s A. 2013 Alzheimer’s disease facts and figures. Alzheimers Dem. 2013;9(2):208–45. - PubMed
-
- Lahiri DK, Farlow MR, Sambamurti K, Greig NH, Giacobini E, Schneider LS. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer’s disease. Curr Drug Targets. 2003;4(2):97–112. - PubMed
-
- Wozniak MA, Frost AL, Itzhaki RF. The helicase-primase inhibitor BAY 57-1293 reduces the Alzheimer’s disease-related molecules induced by herpes simplex virus type 1. Antiviral Res. 2013;99(3):401–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
