

Acetate dependence of tumors
- PMID: 25525877
- PMCID: PMC4272450
- DOI: 10.1016/j.cell.2014.11.020
Acetate dependence of tumors
Abstract
Acetyl-CoA represents a central node of carbon metabolism that plays a key role in bioenergetics, cell proliferation, and the regulation of gene expression. Highly glycolytic or hypoxic tumors must produce sufficient quantities of this metabolite to support cell growth and survival under nutrient-limiting conditions. Here, we show that the nucleocytosolic acetyl-CoA synthetase enzyme, ACSS2, supplies a key source of acetyl-CoA for tumors by capturing acetate as a carbon source. Despite exhibiting no gross deficits in growth or development, adult mice lacking ACSS2 exhibit a significant reduction in tumor burden in two different models of hepatocellular carcinoma. ACSS2 is expressed in a large proportion of human tumors, and its activity is responsible for the majority of cellular acetate uptake into both lipids and histones. These observations may qualify ACSS2 as a targetable metabolic vulnerability of a wide spectrum of tumors.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
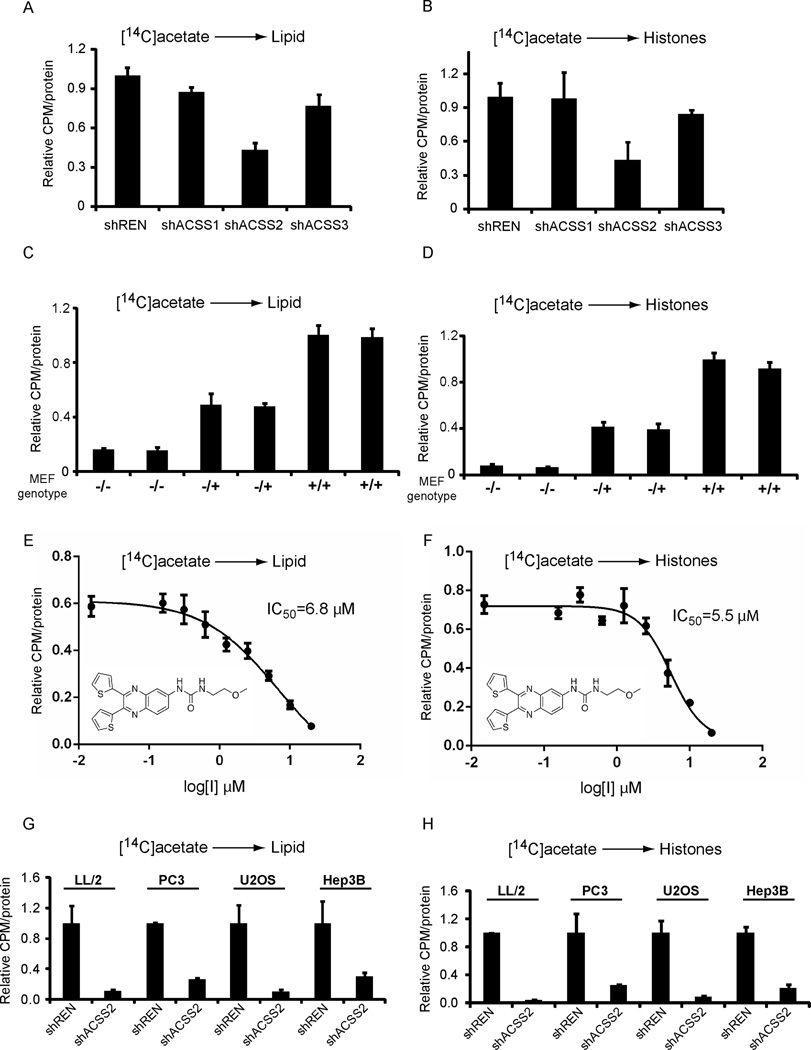
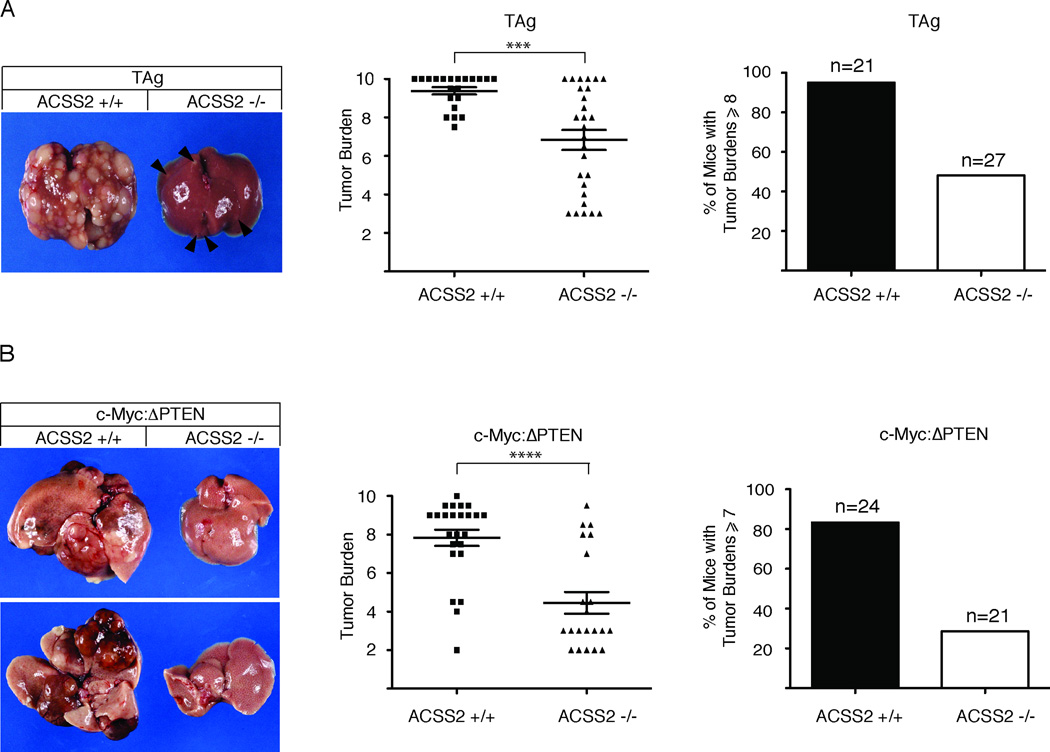
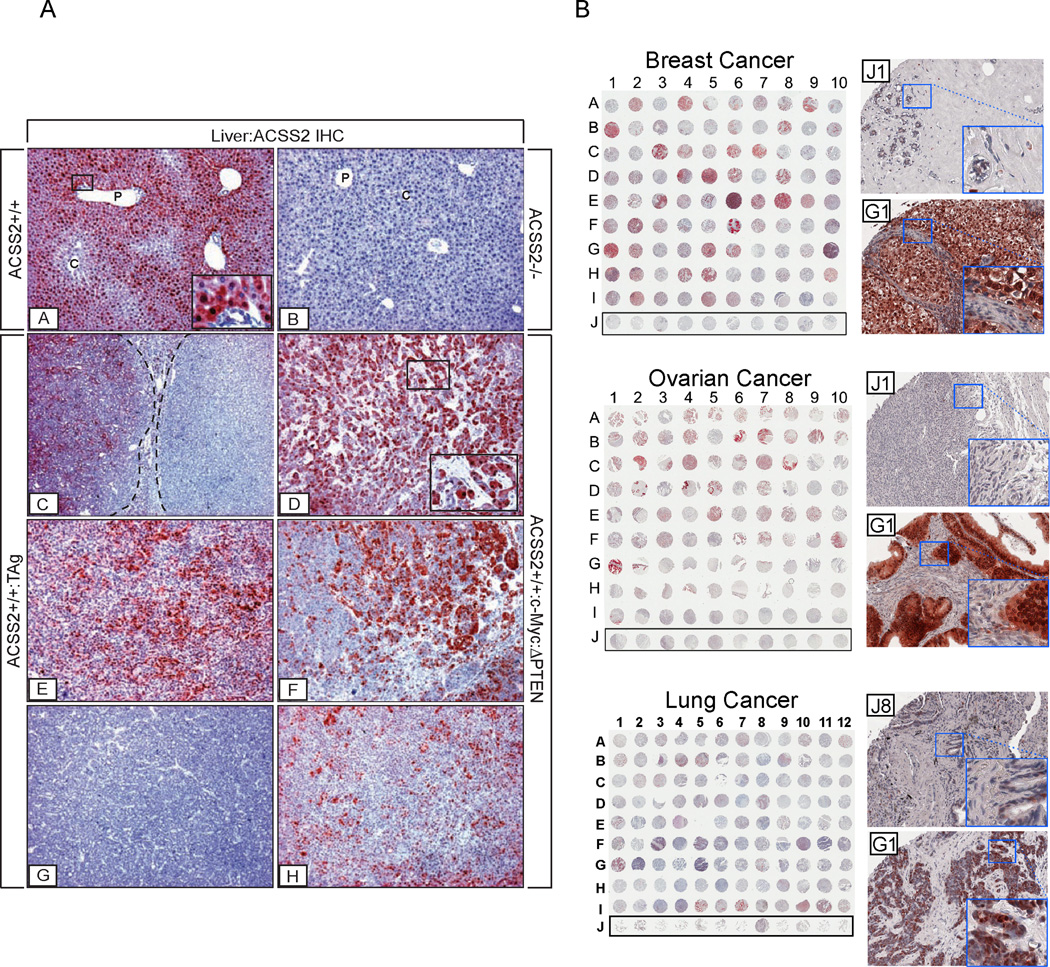
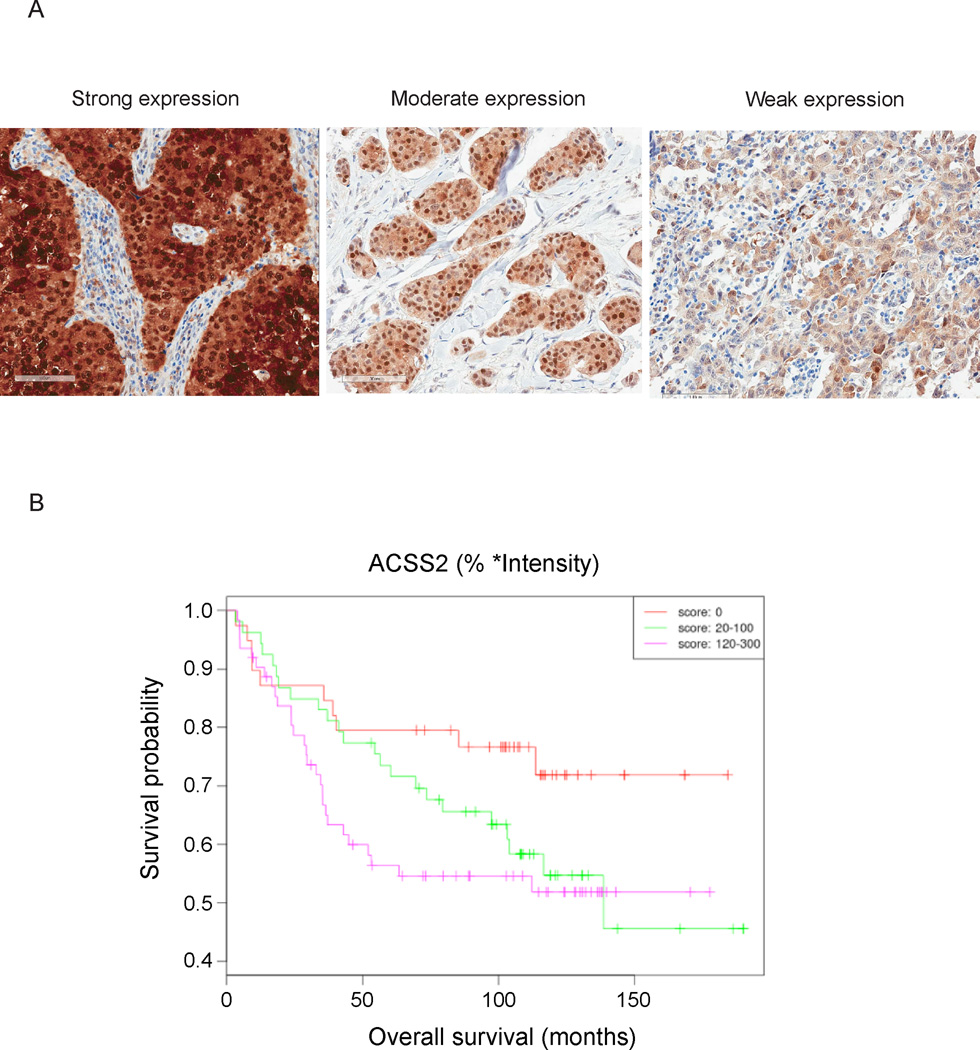
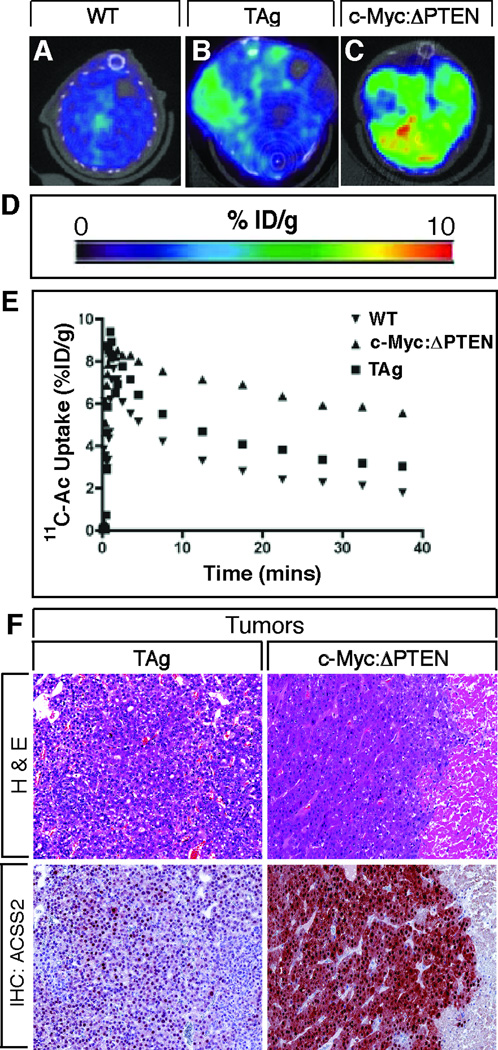
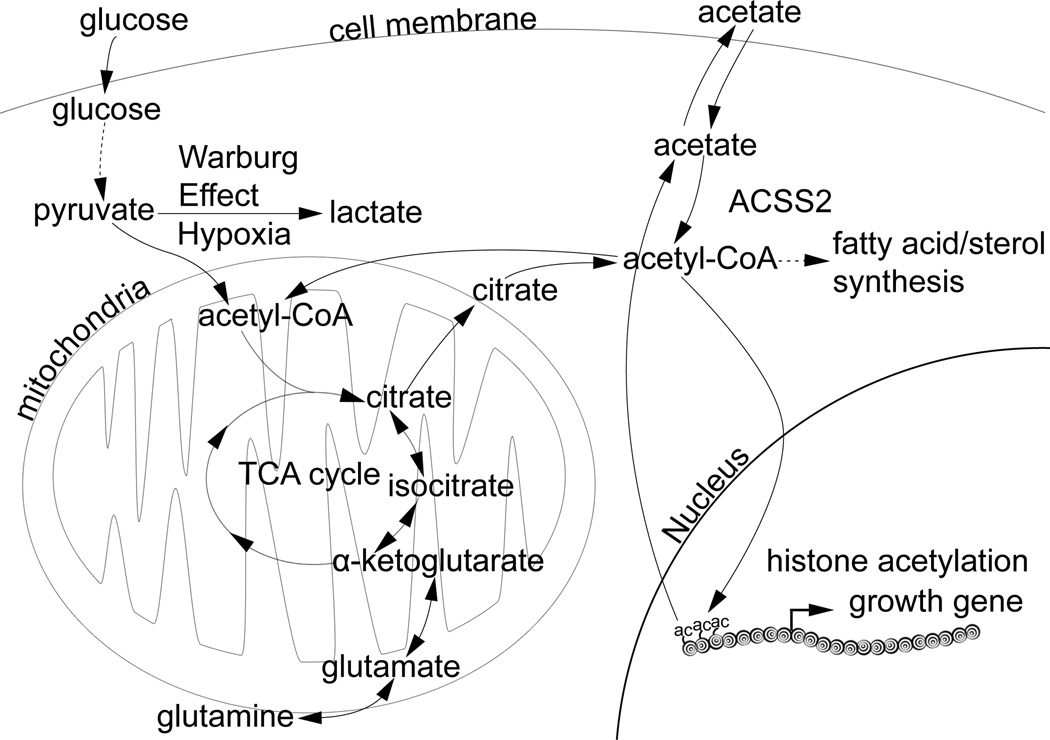
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
