

Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers
- PMID: 25526364
- PMCID: PMC4272259
- DOI: 10.1371/journal.pone.0114263
Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers
Abstract
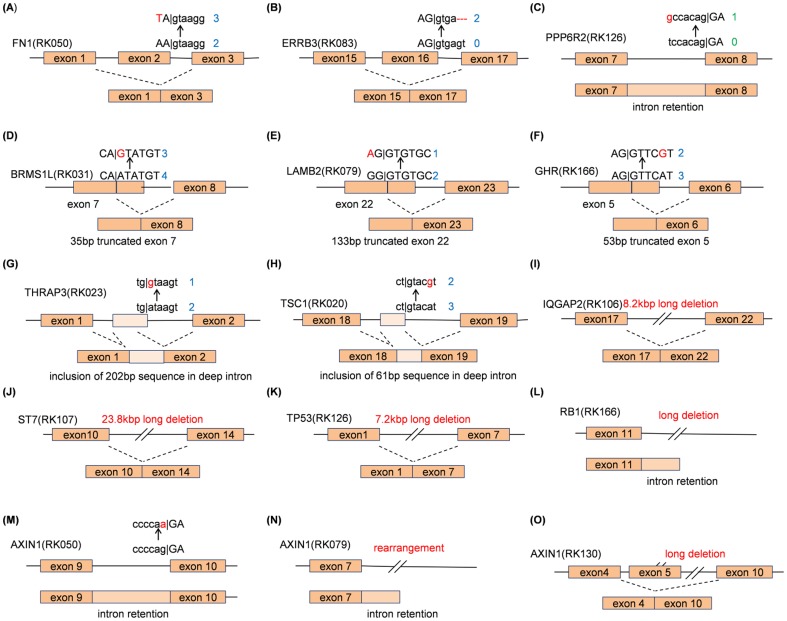
Recent studies applying high-throughput sequencing technologies have identified several recurrently mutated genes and pathways in multiple cancer genomes. However, transcriptional consequences from these genomic alterations in cancer genome remain unclear. In this study, we performed integrated and comparative analyses of whole genomes and transcriptomes of 22 hepatitis B virus (HBV)-related hepatocellular carcinomas (HCCs) and their matched controls. Comparison of whole genome sequence (WGS) and RNA-Seq revealed much evidence that various types of genomic mutations triggered diverse transcriptional changes. Not only splice-site mutations, but also silent mutations in coding regions, deep intronic mutations and structural changes caused splicing aberrations. HBV integrations generated diverse patterns of virus-human fusion transcripts depending on affected gene, such as TERT, CDK15, FN1 and MLL4. Structural variations could drive over-expression of genes such as WNT ligands, with/without creating gene fusions. Furthermore, by taking account of genomic mutations causing transcriptional aberrations, we could improve the sensitivity of deleterious mutation detection in known cancer driver genes (TP53, AXIN1, ARID2, RPS6KA3), and identified recurrent disruptions in putative cancer driver genes such as HNF4A, CPS1, TSC1 and THRAP3 in HCCs. These findings indicate genomic alterations in cancer genome have diverse transcriptomic effects, and integrated analysis of WGS and RNA-Seq can facilitate the interpretation of a large number of genomic alterations detected in cancer genome.
Conflict of interest statement
Figures





References
-
- El-Serag HB (2011) Hepatocellular carcinoma. N Engl J Med 365:1118–1127. - PubMed
-
- Farazi PA, DePinho RA (2006) Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 6:674–687. - PubMed
-
- Meyerson M, Gabriel S, Getz G (2010) Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet 11:685–696. - PubMed
-
- Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, et al. (2012) Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 44:760–764. - PubMed
-
- Huang J, Deng Q, Wang Q, Li KY, Dai JH, et al. (2012) Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet 44:1117–1121. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
