

PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402
- PMID: 25527497
- PMCID: PMC4317039
- DOI: 10.1074/jbc.M114.620906
PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402
Abstract
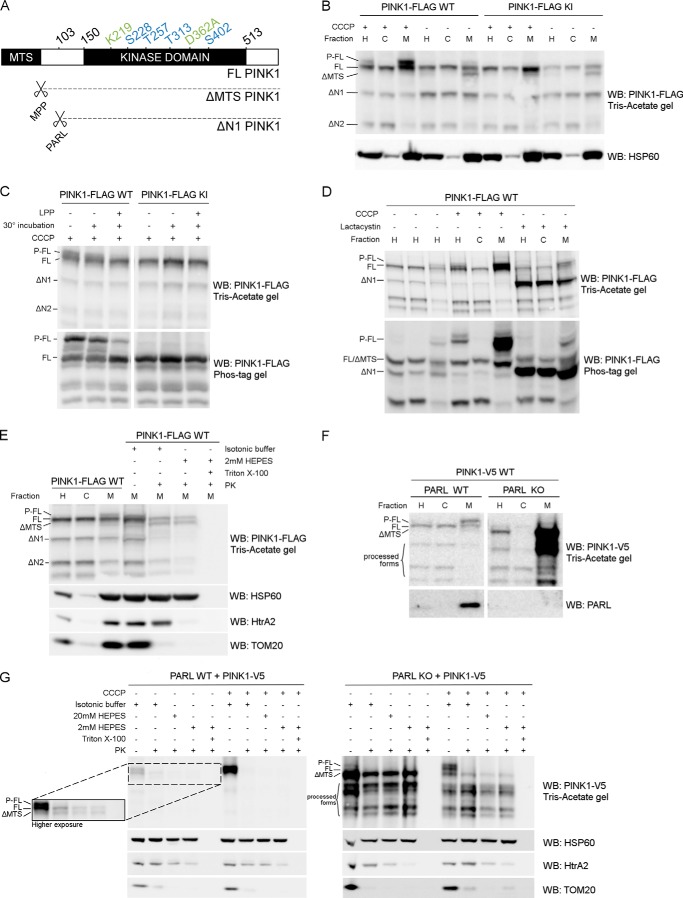
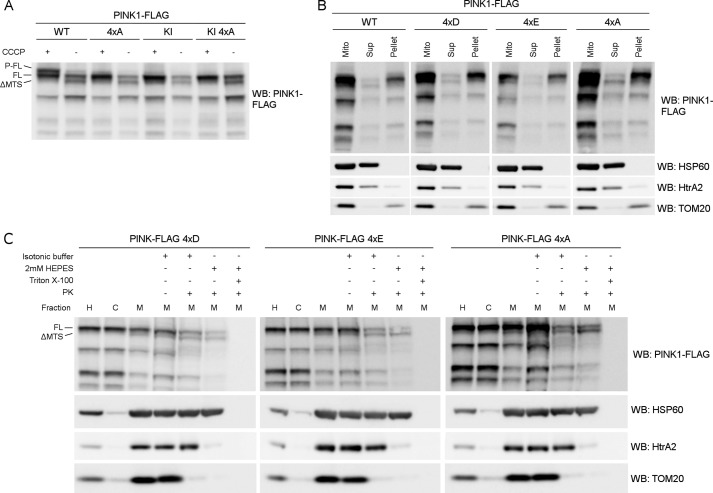
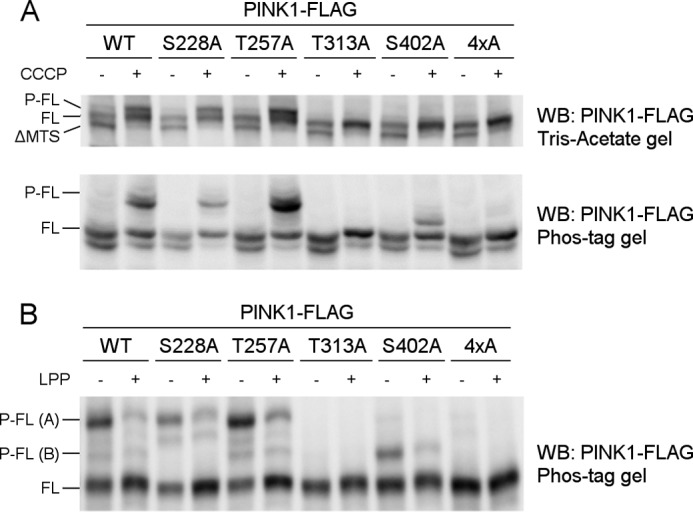
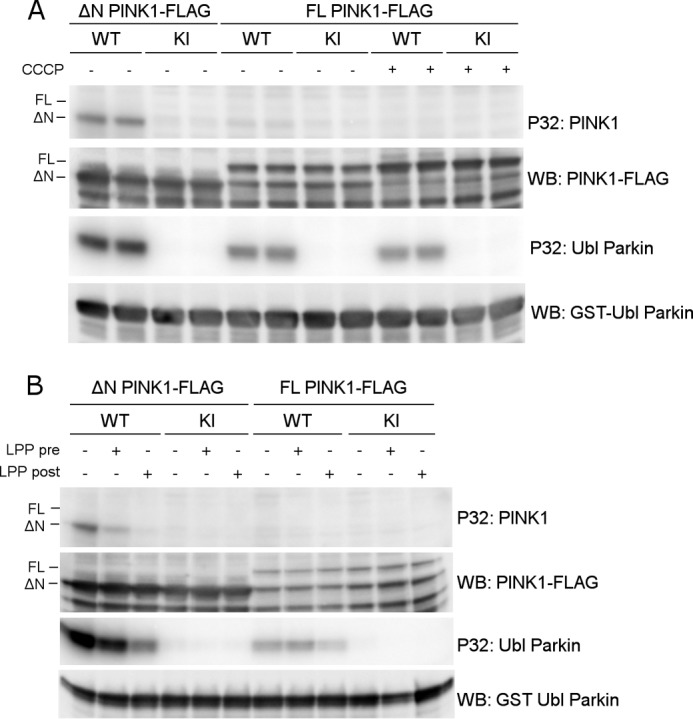
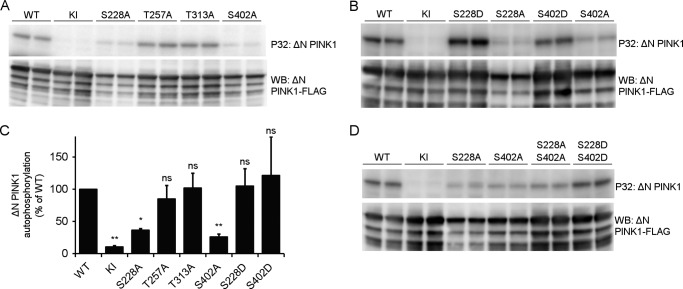
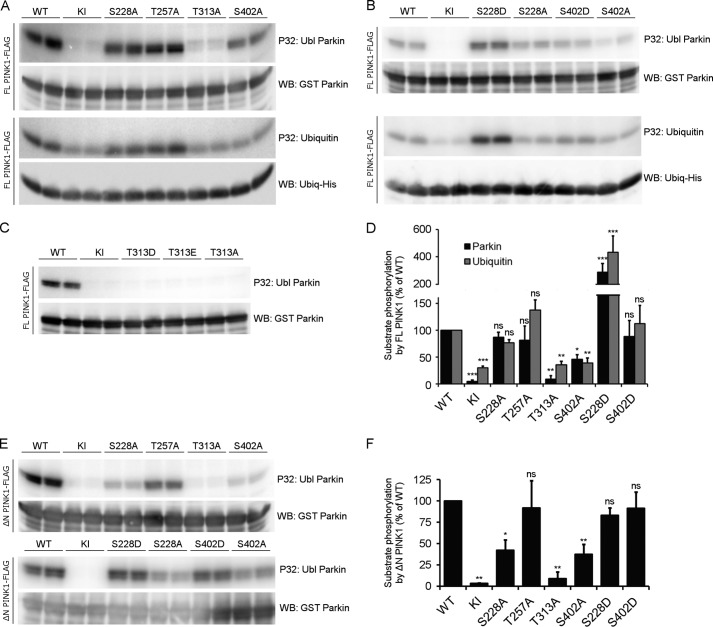
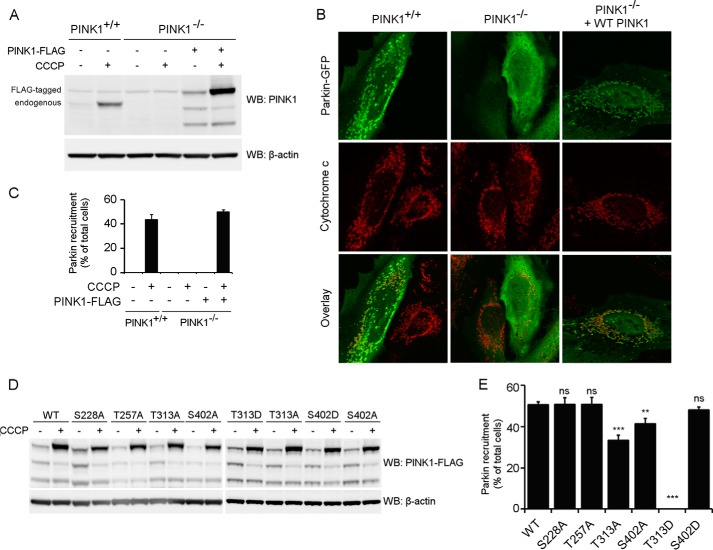
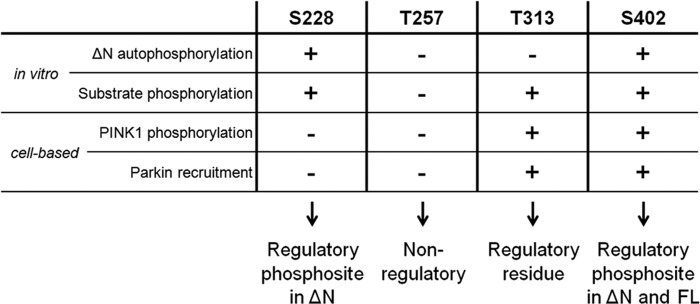
Mutations in the PINK1 gene cause early-onset recessive Parkinson disease. PINK1 is a mitochondrially targeted kinase that regulates multiple aspects of mitochondrial biology, from oxidative phosphorylation to mitochondrial clearance. PINK1 itself is also phosphorylated, and this might be linked to the regulation of its multiple activities. Here we systematically analyze four previously identified phosphorylation sites in PINK1 for their role in autophosphorylation, substrate phosphorylation, and mitophagy. Our data indicate that two of these sites, Ser-228 and Ser-402, are autophosphorylated on truncated PINK1 but not on full-length PINK1, suggesting that the N terminus has an inhibitory effect on phosphorylation. We furthermore establish that phosphorylation of these PINK1 residues regulates the phosphorylation of the substrates Parkin and Ubiquitin. Especially Ser-402 phosphorylation appears to be important for PINK1 function because it is involved in Parkin recruitment and the induction of mitophagy. Finally, we identify Thr-313 as a residue that is critical for PINK1 catalytic activity, but, in contrast to previous reports, we find no evidence that this activity is regulated by phosphorylation. These data clarify the regulation of PINK1 through multisite phosphorylation.
Keywords: Mitochondria; Neurodegenerative Disease; PTEN-induced Putative Kinase 1 (PINK1); Parkinson Disease; Posttranslational Modification (PTM); Protein Phosphorylation; Serine/Threonine Protein Kinase.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 - PubMed
-
- Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J.-W., Yang L., Beal M. F., Vogel H., Lu B. (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U.S.A. 103, 10793–10798 - PMC - PubMed
-
- Exner N., Treske B., Paquet D., Holmström K., Schiesling C., Gispert S., Carballo-Carbajal I., Berg D., Hoepken H.-H., Gasser T., Krüger R., Winklhofer K. F., Vogel F., Reichert A. S., Auburger G., Kahle P. J., Schmid B., Haass C. (2007) Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 27, 12413–12418 - PMC - PubMed
-
- Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
