

G protein-coupled estrogen receptor protects from atherosclerosis
- PMID: 25532911
- PMCID: PMC4274506
- DOI: 10.1038/srep07564
G protein-coupled estrogen receptor protects from atherosclerosis
Erratum in
-
Erratum: G Protein-coupled Estrogen Receptor Protects from Atherosclerosis.Sci Rep. 2015 Sep 16;5:13510. doi: 10.1038/srep13510. Sci Rep. 2015. PMID: 26373621 Free PMC article. No abstract available.
Abstract
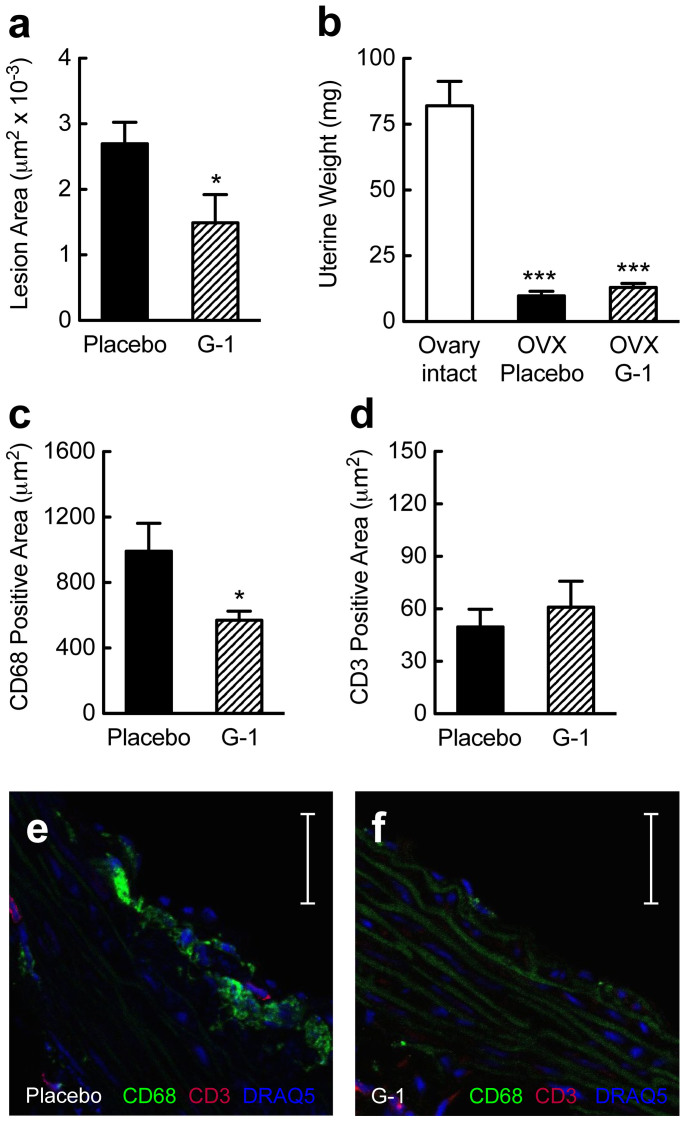
Coronary atherosclerosis and myocardial infarction in postmenopausal women have been linked to inflammation and reduced nitric oxide (NO) formation. Natural estrogen exerts protective effects on both processes, yet also displays uterotrophic activity. Here, we used genetic and pharmacologic approaches to investigate the role of the G protein-coupled estrogen receptor (GPER) in atherosclerosis. In ovary-intact mice, deletion of gper increased atherosclerosis progression, total and LDL cholesterol levels and inflammation while reducing vascular NO bioactivity, effects that were in some cases aggravated by surgical menopause. In human endothelial cells, GPER was expressed on intracellular membranes and mediated eNOS activation and NO formation, partially accounting for estrogen-mediated effects. Chronic treatment with G-1, a synthetic, highly selective small molecule agonist of GPER, reduced postmenopausal atherosclerosis and inflammation without uterotrophic effects. In summary, this study reveals an atheroprotective function of GPER and introduces selective GPER activation as a novel therapeutic approach to inhibit postmenopausal atherosclerosis and inflammation in the absence of uterotrophic activity.
Figures
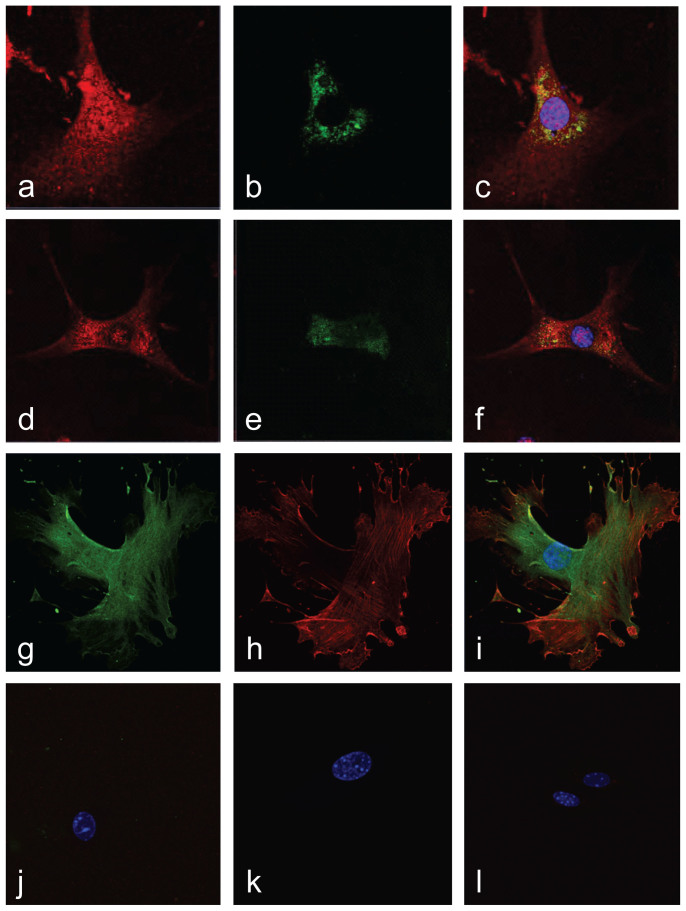
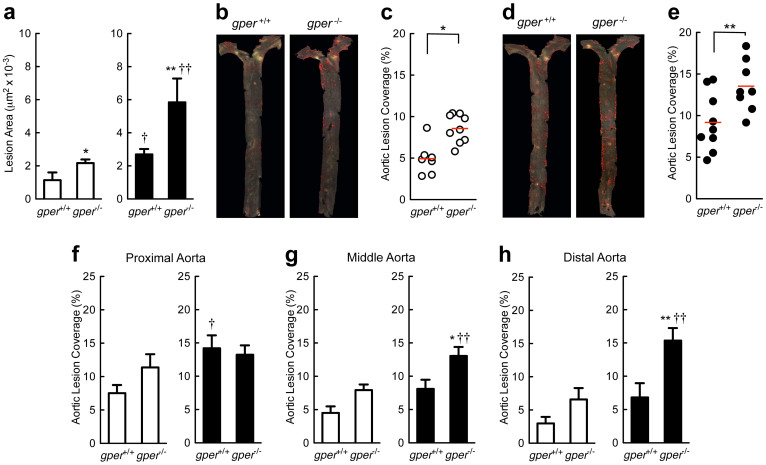
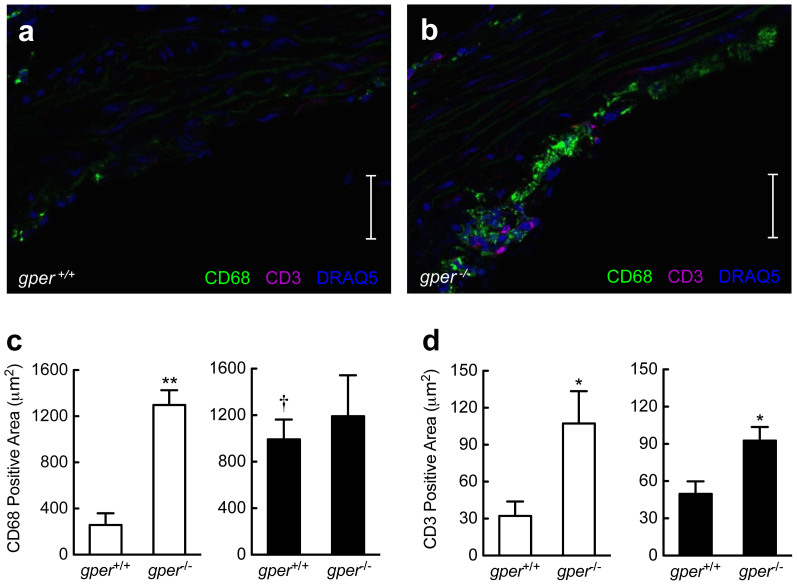
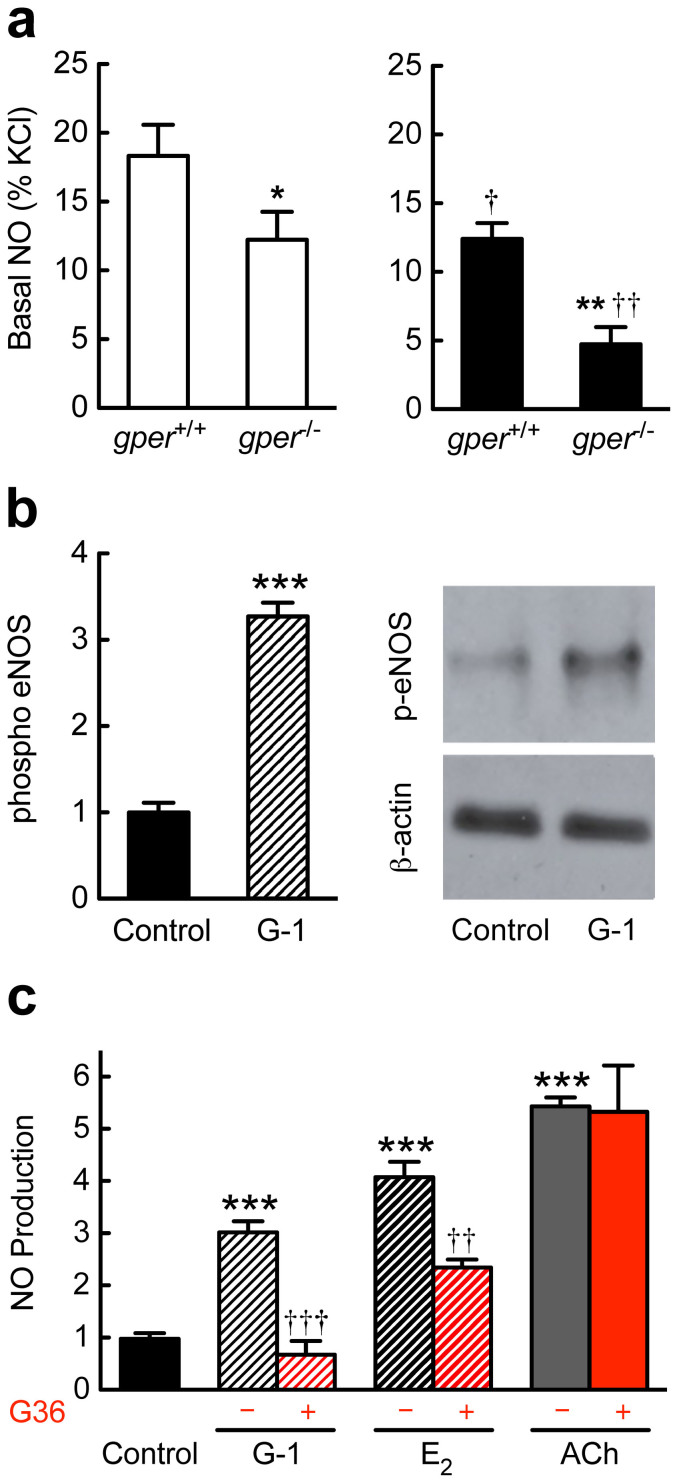
References
-
- Turgeon J. L., McDonnell D. P., Martin K. A. & Wise P. M. Hormone therapy: physiological complexity belies therapeutic simplicity. Science 304, 1269–1273 (2004). - PubMed
-
- Hodis H. N. & Mack W. J. Hormone replacement therapy and the association with coronary heart disease and overall mortality: clinical application of the timing hypothesis. J Steroid Biochem Mol Biol 142, 68–75 (2014). - PubMed
-
- Phillips G. B., Pinkernell B. H. & Jing T. Y. Relationship between serum sex hormones and coronary artery disease in postmenopausal women. Arterioscler Thromb Vasc Biol 17, 695–701 (1997). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
