

Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes
- PMID: 25535334
- PMCID: PMC4323486
- DOI: 10.1128/MCB.01109-14
Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes
Abstract
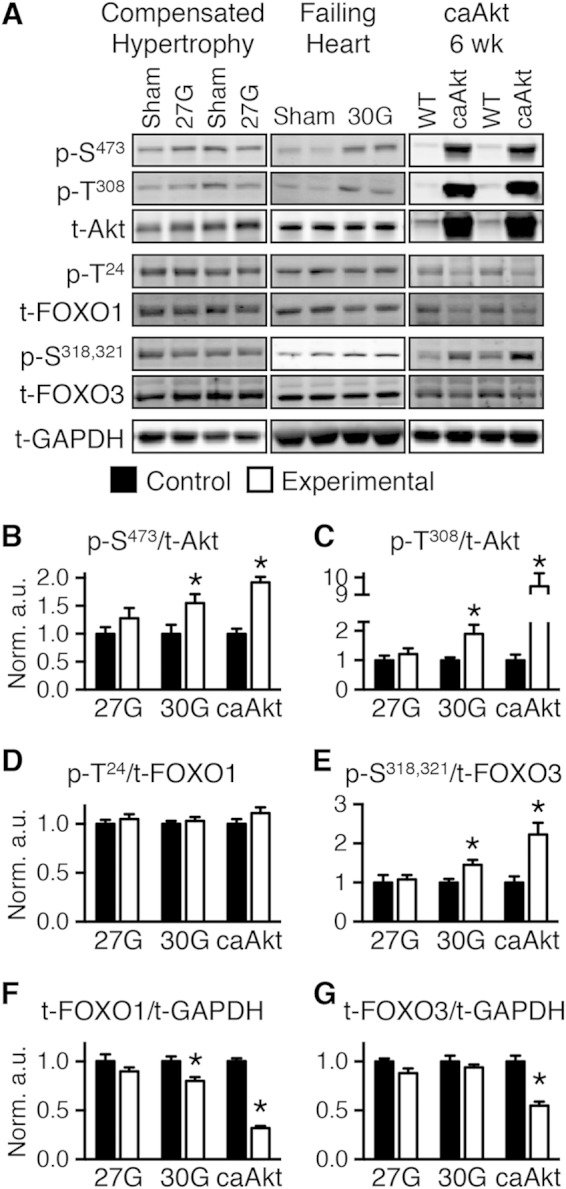
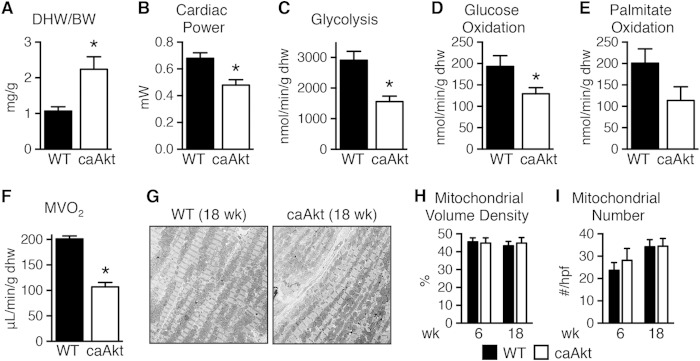
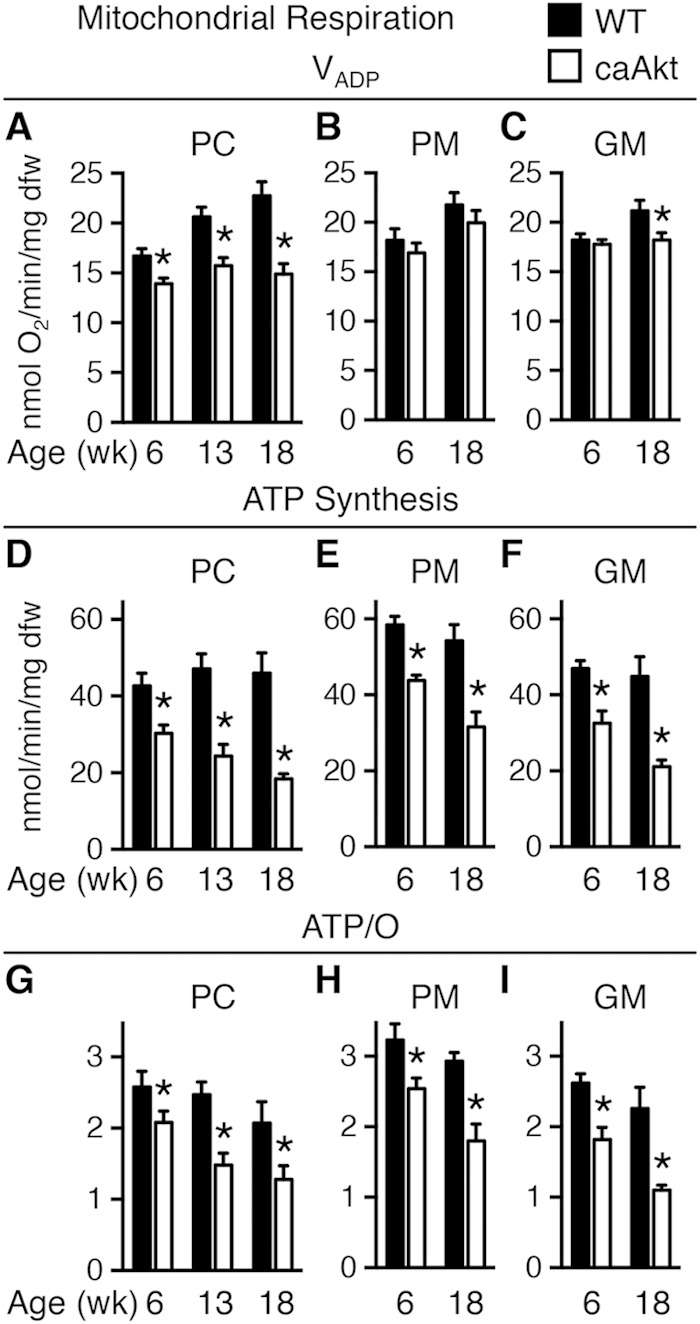
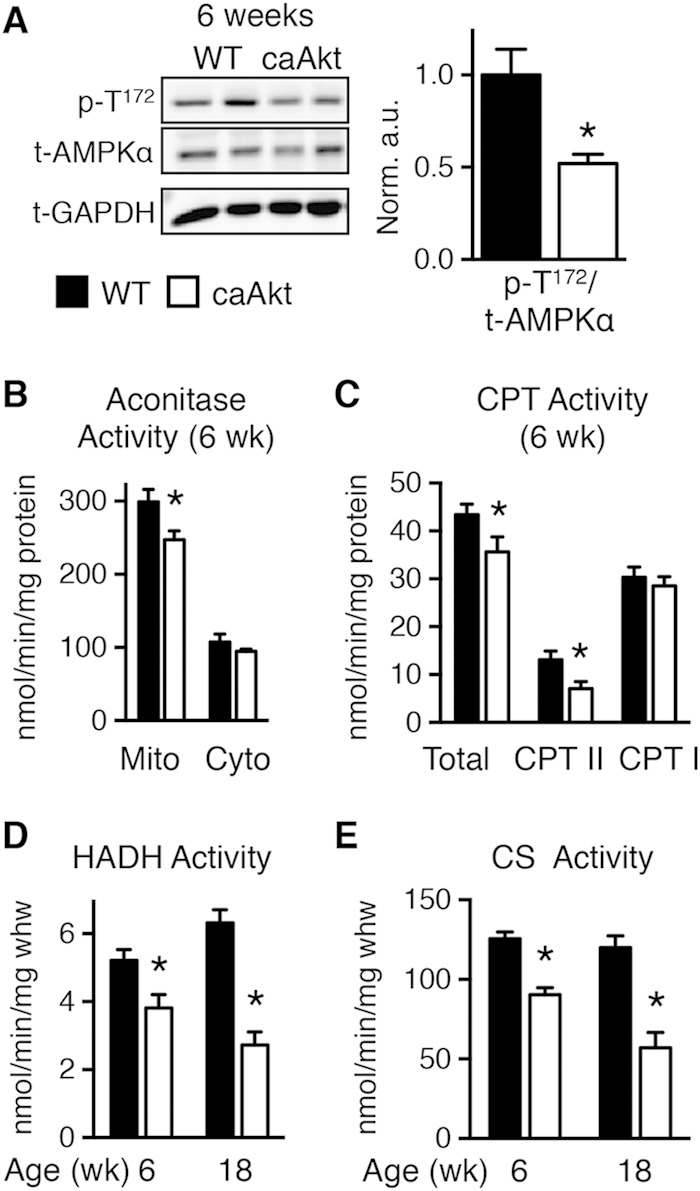
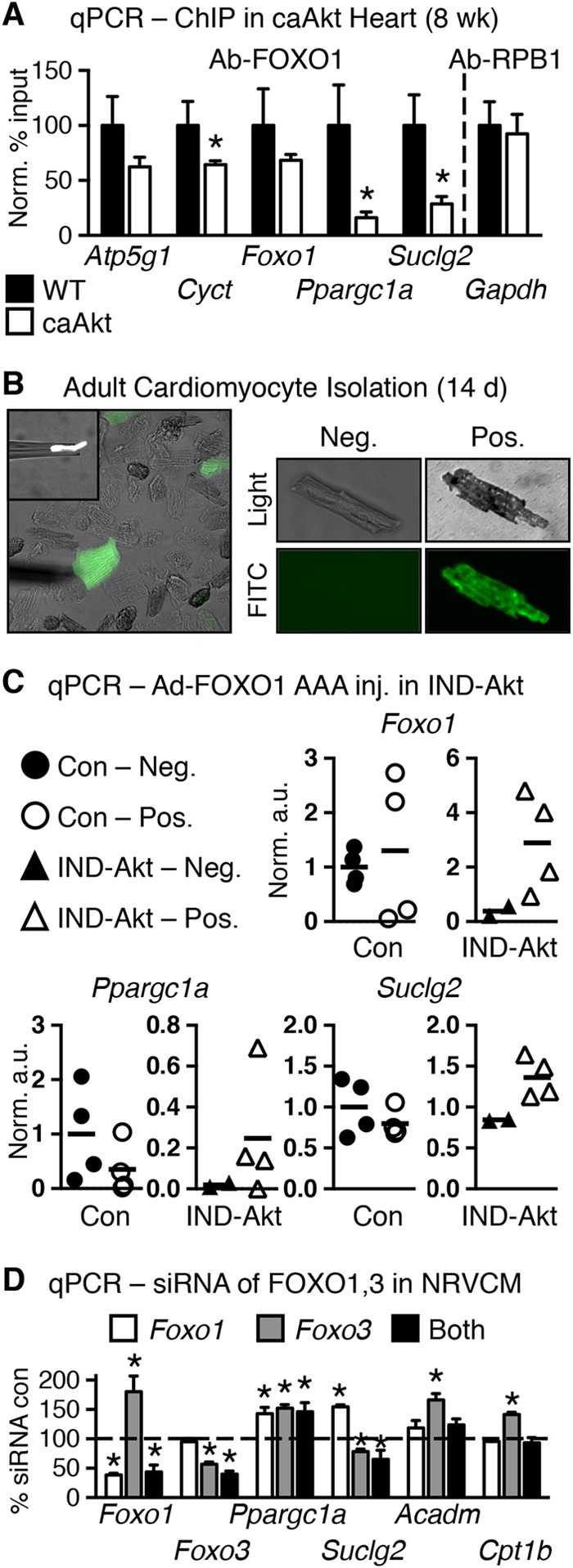
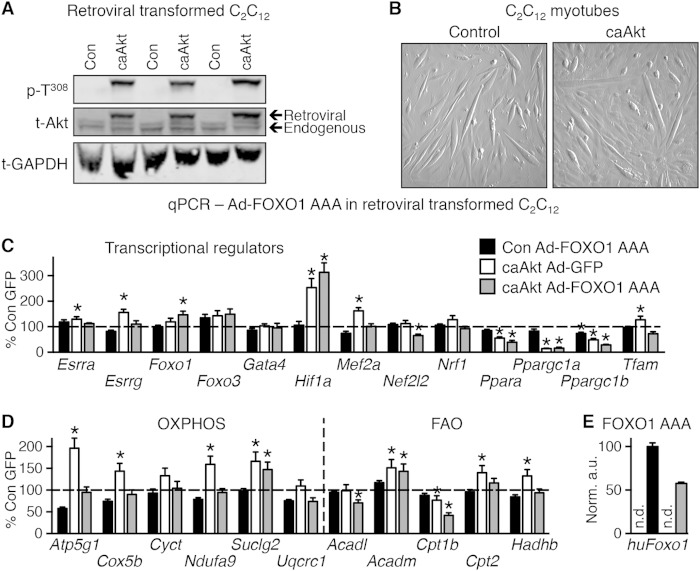
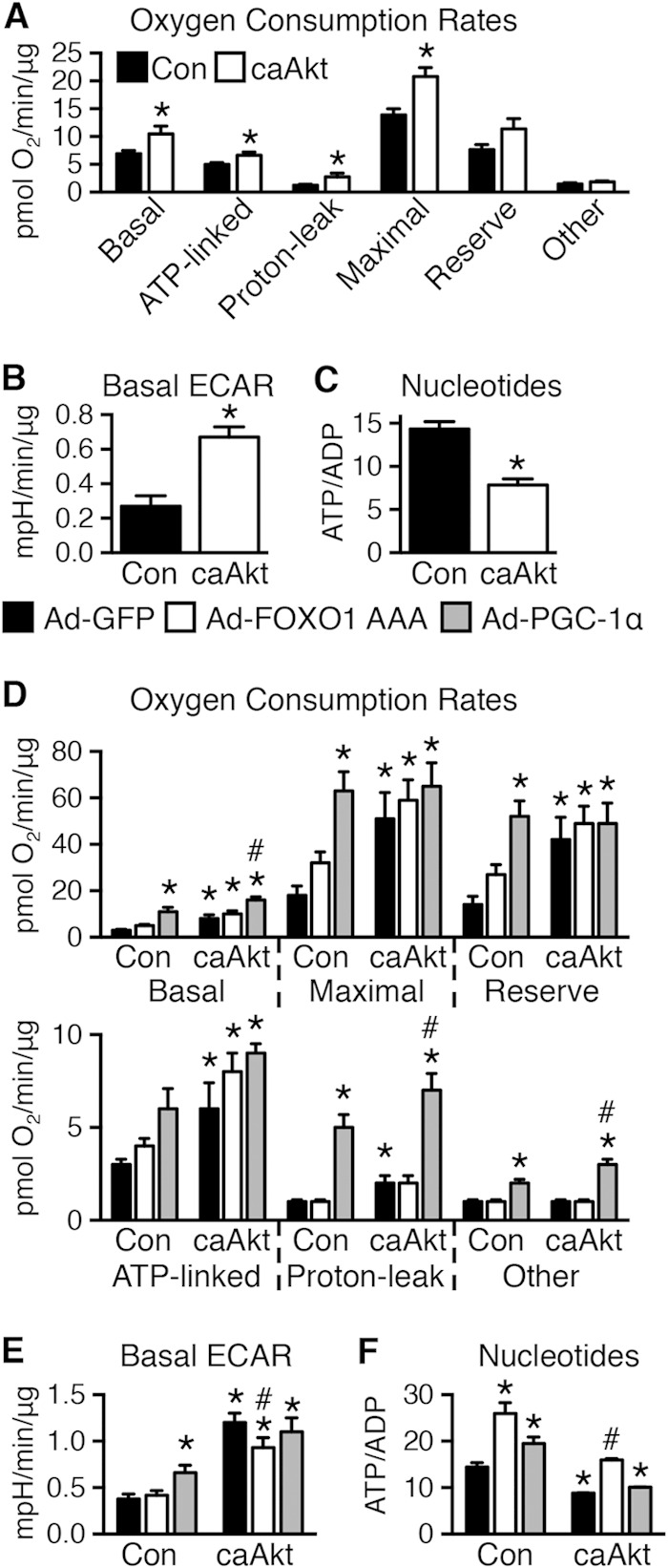
Sustained Akt activation induces cardiac hypertrophy (LVH), which may lead to heart failure. This study tested the hypothesis that Akt activation contributes to mitochondrial dysfunction in pathological LVH. Akt activation induced LVH and progressive repression of mitochondrial fatty acid oxidation (FAO) pathways. Preventing LVH by inhibiting mTOR failed to prevent the decline in mitochondrial function, but glucose utilization was maintained. Akt activation represses expression of mitochondrial regulatory, FAO, and oxidative phosphorylation genes in vivo that correlate with the duration of Akt activation in part by reducing FOXO-mediated transcriptional activation of mitochondrion-targeted nuclear genes in concert with reduced signaling via peroxisome proliferator-activated receptor α (PPARα)/PGC-1α and other transcriptional regulators. In cultured myocytes, Akt activation disrupted mitochondrial bioenergetics, which could be partially reversed by maintaining nuclear FOXO but not by increasing PGC-1α. Thus, although short-term Akt activation may be cardioprotective during ischemia by reducing mitochondrial metabolism and increasing glycolysis, long-term Akt activation in the adult heart contributes to pathological LVH in part by reducing mitochondrial oxidative capacity.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, Bugger H, Buchanan J, Wang X, Moreira AB, Doenst T, Medina-Gomez G, Litwin SE, Lelliott CJ, Vidal-Puig A, Abel ED. 2011. PGC-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res 109:783–793. doi: 10.1161/CIRCRESAHA.111.243964. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous